











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
La distrofia muscular de Duchenne y Becker (DMD/DMB) son enfermedades neuromusculares con herencia recesiva ligada al cromosoma X ocasionada por mutaciones en el gen de la distrofina. Estas mutaciones generan deficiencia de dicha proteína citoesquelética con la consecuente pérdida progresiva de la funcionalidad de las fibras musculares 1. La sintomatología puede presentarse desde los 2 a 3 años, aunque pueden estar precedidos por retraso psicomotor. En el niño, clínicamente se caracteriza por debilidad progresiva, dificultad para ponerse de pie (signo de Gowers), dificultad para subir escaleras y caminar de puntas 2. Mazzone y col., reportaron que un 3 % pierde la deambulación cada año. En estadios más avanzados se puede presentar escoliosis (3.9 % - 52.1 %), cardiomiopatía (21.2 % - 57.3 %), dificultad respiratoria de tipo ventilatorio que requiere apoyo asistido (0 % - 22 %) entre otras complicaciones (3, 4).
La DMD es la distrofia muscular más común afectando a 1 de cada 3800-6300 varones nacidos vivos 5. Su prevalencia mundial es de aproximadamente 0.5 por cada 100 000 varones 6. Pocos estudios han estudiado la prevalencia de DMD/DMB en la población masculina de todas las edades. Un estudio poblacional realizado en el norte del Reino Unido halló 124 casos confirmados con pruebas genéticas y biopsia muscular reportando una prevalencia puntual de 8,3 por cada 100.000 varones 7. Además, la sobrevida de pacientes con DMD varía entre 24 a 26 años según estudios realizados en Italia, Francia y Alemania 3. En el Perú, los datos acerca de la prevalencia de DMD son limitados, con un estudio reportando un porcentaje de 0.49 % en una muestra de 7829 en una población hospitalaria, siendo este valor ligeramente mayor a la prevalencia mundial 8.
Debido a que la DMD es una enfermedad de baja prevalencia, pero muy discapacitante, se ha considerado como una enfermedad poco frecuente por el “Genetic and Rare Diseases Information Center” del “National Institutes of Health” (NIH) y las terapias probables están dentro de la lista de medicamentos huérfanos de la “Food and Drug Administration” (FDA) de los EE. UU. En el Perú la Ley N.º 29698 reconoce y declara de interés nacional el diagnóstico y tratamiento de este tipo de enfermedades 5. Considerando la distrofia muscular de Duchenne como la distrofia muscular más prevalente a nivel mundial, la Sociedad Peruana de Neurología priorizó elaborar esta Guía de Práctica Clínica (GPC)
OBJETIVO Y POBLACIÓN
Contribuir a la identificación temprana de pacientes con sospecha de DMD/DMB de todas las edades y brindar recomendaciones para el tamizaje, diagnóstico y tratamiento de esta patología.
USUARIOS Y ÁMBITO DE LA GPC
La presente GPC está dirigida a los profesionales de la salud vinculados con la atención de pacientes con DMD y DMB. El ámbito de aplicación de la GPC son establecimientos de salud públicos y privados involucrados en el manejo de niños, adolescentes y adultos con enfermedades neuromusculares, según corresponda su nivel de complejidad, disponibilidad de profesionales de salud, técnicas diagnósticas y medicamentos.
MÉTODOS
Conformación del grupo elaborador de la guía local (GEG): Se conformó un GEG que incluyó neurólogos, neuropediatras, genetistas, metodólogos, expertos en calidad de servicios de salud y representantes de pacientes. Solo una integrante declaró potenciales conflictos de intereses que no fueron considerados relevantes para el desarrollo de la presente GPC por el GEG-Local. El resto de integrantes declaró no tenerlos.
Formulación de preguntas: El GEG-Local formuló un listado de preguntas clínicas en formato PICO (Population, Intervention, Comparator, Outcome) (Tabla 1).

Búsqueda, selección y síntesis de la evidencia: Para cada pregunta PICO se realizó una búsqueda sistemática (BS) de GPC basadas en revisiones sistemáticas (RSs) actualizadas (periodo 2015-2020), de no encontrarse se buscó y/o actualizaron RSs o estudios primarios de acuerdo con cada PICO. Se realizaron síntesis de evidencia numérica (metaanálisis, MA) o narrativa por desenlace para cada PICO (Figura 2).
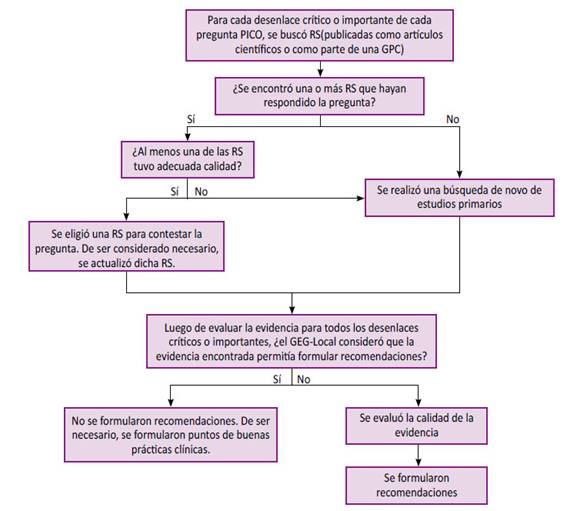
Figura 2 Flujograma de selección de la evidencia para la formulación de recomendaciones y puntos de buenas prácticas clínicas
Evaluación de calidad: Cada RS y estudio primario fue evaluada en calidad con su herramienta validada correspondiente, AMSTAR 2 para RS 9, la herramienta Cochrane para los Ensayos clínicos aleatorizados (ECA) 10, Newcastle-Ottawa para estudios observacionales 11, QUADAS-2 para estudios diagnósticos 12 y la herramienta propuesta por Murad y col. para estudios descriptivos que no evaluaron una intervención ni consideraron grupo comparador 13.
Evaluación de la certeza de la evidencia: La certeza global de la evidencia para cada PICO pudo ser alta, moderada, baja, y muy baja, según el sistema GRADE (Grading of Recommendations Assessment, Development and Evaluation). Esta se basa en 9 aspectos: tipo de estudio, riesgo de sesgo, inconsistencia, evidencia indirecta, imprecisión, sesgo de publicación, tamaño de efecto, relación dosis-respuesta, y efecto de confusores (los tres últimos aspectos son evaluados en estudios observacionales) 14.
Formulación de las recomendaciones: El GEG revisó la evidencia seleccionada para cada pregunta clínica en reuniones periódicas, y formuló recomendaciones fuertes o condicionales por consenso o por mayoría simple, usando el sistema GRADE 15. Para ello, se tuvo en consideración: 1) beneficios y daños de las opciones, 2) valores y preferencias de los pacientes, 3) aceptabilidad por parte de los profesionales de salud, 4) factibilidad y aplicabilidad de las opciones, 5) impacto en la equidad y 6) uso de recursos. Asimismo, el GEG formuló puntos de buenas prácticas clínicas (BPC) y consideraciones de implementación por consenso, cuando no se encontró evidencia suficiente para formular una recomendación.
Revisión por expertos externos: La presente GPC fue revisada por médicos especialistas representantes de otras instituciones y tomadores de decisiones. Asimismo, su versión in-extenso fue enviada por vía electrónica a externos para su revisión temática y metodológica (mencionados en la sección de agradecimientos). El GEG tuvo en cuenta los resultados de estas revisiones para realizar modificaciones y mejoras.
Actualización de la GPC: La presente GPC tiene una validez de tres años. Al acercarse al fin de este período, se procederá a realizar una RS de la literatura para su actualización, luego de la cual se decidirá si se actualiza la presente GPC o se procede a realizar una nueva versión.
El resto de información de referencias, detalles metodológicos y resultados están descritos en la versión extensa de la guía, publicada en la web de la Sociedad Peruana de Neurología (https://www.spneurologia.org.pe)
DESARROLLO DE RECOMENDACIONES
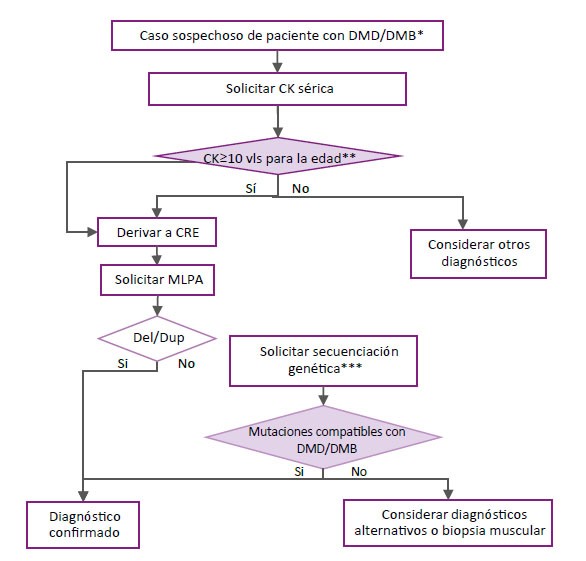
El GEG consideró formular como punto de BPC la siguiente definición de casos de sospecha para pacientes con DMD/DMB: Varón con debilidad muscular progresiva a predominio de miembros inferiores, con o sin antecedente familiar de DMD-DMB y alguno de los siguientes síntomas o signos: pseudohipertrofia de pantorrillas, retraso psicomotor, retraso en la adquisición de la marcha, dificultad para subir o bajar escaleras, incapacidad para incorporarse desde el suelo, trepar sobre su cuerpo para incorporarse (Signo de Gowers), dificultad para caminar (marcha dandineante) o correr y caídas frecuentes.
Pregunta 1. En paciente con sospecha clínica de DMD/DMB, ¿la prueba sérica de creatina quinasa (CK) es útil para el tamizaje diagnóstico?
La degeneración de fibras musculares ocasiona liberación de enzimas intracelulares como la creatina quinasa (CK) en las DMD/ DMB que puedan ser detectadas como biomarcadores séricos de lesión muscular (16, 17). Sin embargo, también puede estar elevada en otras DM 18 y está influenciado por factores como el nivel de actividad y masa muscular 19. El GEG consideró importante determinar la utilidad del CK para el tamizaje (sensibilidad) de DMD/DMB (1, 20).

De la evidencia a la decisión:
Para determinar el rendimiento de la prueba sérica de CK se incluyeron 9 estudios. Dos de ellos, con un diseño de prueba diagnóstica y 7 de ellos con un diseño tipo caso-control. El meta- análisis (MA) de los 9 estudios observacionales (n=1745) estimó una alta sensibilidad 98 % (IC95 %: 95 % a 99 %) y especificidad 82 % (IC95%: 33 % a 98 %) con elevada heterogeneidad, I2 de 99.03 % (IC95 %: 98.81-99.24) y 98.88 % (IC95 % (98.62-99.14) respectivamente. El área Bajo la Curva (ABC) fue de 0.99 (IC95 %: 0.97 a 0.99).
El conjunto de evidencia presentó elevado riesgo de sesgo e inconsistencia, certeza global moderada a muy baja. Sin embargo, el GEG formuló una recomendación fuerte a favor del CK para el tamizaje de pacientes con DMD, por tener alta sensibilidad, ser aceptable y aplicable, además de factible y de bajo costo.
Pregunta 2: En paciente con sospecha clínica y de laboratorio (CK) de DMD/DMB, ¿la prueba molecular “MLPA” es útil para establecer el diagnóstico de DMD/DMB?
El MLPA es una técnica basada en sondas genéticas para la detección de la deleción y duplicación de genes, especialmente para aquellas deleciones de tamaño mediano que no pueden ser detectadas por el PCR o las técnicas citogenéticas 21 y que son aplicadas para algunas enfermedades como las deleciones BRCA1 y BRCA2 asociadas al cáncer de mama familiar 22 o para el gen DMD asociado a la distrofia muscular de Duchenne. Algunas limitaciones son su baja capacidad de detección en deleciones con polimorfismos o mutaciones de una sola base 21. El GEG consideró determinar su exactitud para establecer el diagnóstico de DMD/DMB.
De la evidencia a la decisión:
Se encontraron 10 estudios observacionales, de diseño transversal y de cohorte. El MA fue de 10 estudios observacionales (n=3535) estimó una sensibilidad de 0.80 (IC-95 %: 0.76-0.84), con alta heterogeneidad (I2: 86.48); y especificidad de 0.93 (IC-95 %: 0.87-0.96). con baja heterogeneidad (I2: 47.27). El ABC fue de 0.94 (IC-95 %: 0.92-0.96).
El conjunto de evidencia presentó, con elevada inconsistencia, certeza global de moderada a baja. Sin embargo, tiene buena sensibilidad y alta especificidad, es aceptable, aplicable y factible, por lo cual el GEG formuló una recomendación condicional a favor del uso de la prueba de MLPA para el diagnóstico de pacientes con DMD.

Pregunta 3: En paciente con sospecha clínica y de laboratorio (CK) de DMD negativos en la prueba “MLPA”, ¿la prueba de secuenciamiento es útil para establecer en el diagnóstico de DMD/DMB?
El NGS corresponde a un conjunto de técnicas con capacidad de evaluar millones de cadenas modelo en paralelo, gracias a su distribución espacial 23. Su aparición hace poco más de 10 años significó una reducción drástica en tiempo y costo, y simplificó el flujo diagnóstico en enfermedades raras, incluyendo neuromusculares como la DMD 24. Esta es capaz de detectar tanto mutaciones grandes como pequeñas. Sin embargo, debido a su costo, su uso se ha limitado principalmente a estas últimas 25. Debido a la presencia de polimorfismos en DMD, es necesario añadir una técnica que pueda superar la baja tasa de detección de estos por MLPA. El GEG consideró que determinar la utilidad de esta prueba para el diagnóstico de DMD es un complemento importante.
De la evidencia a la decisión:
Para responder a esta pregunta PICO se utilizó la tasa de detección (TD) del NGS, definida como número de casos donde se obtuvo el diagnóstico de DMD/DMB sobre el número de casos evaluados. De los 14 estudios observacionales seleccionados, de diseño transversal y de cohorte, el MA (n=881) de la tasa de detección fue de 73.17 (IC95 %: 60.46 - 85.88) con alta heterogeneidad (I2:95.96 %). Finalmente se evaluó la TD del algoritmo combinado de MLPA-NGS. Se seleccionaron 10 estudios observacionales, de diseño transversal y de cohorte, su MA (n= 3958) estimó una tasa de detección de 94.73 (IC95 %: 92.34 - 97.11) con alta heterogeneidad (I2: 95.41 %).
El conjunto de evidencia presentó, con elevada inconsistencia, certeza global baja. Sin embargo, genera de regular a alta tasa de detección, es aceptable, aplicable y factible, por lo cual el GEG formuló una recomendación fuerte a favor del uso de secuenciamiento y del algoritmo MLPA/NGS para el diagnóstico de pacientes con DMD/DMB

Pregunta 4: En pacientes con diagnóstico de DMD, ¿es eficaz y seguro el tratamiento con glucocorticoides?
Hasta la actualidad, la fisioterapia y el tratamiento con glucocorticoides siguen siendo los pilares del tratamiento de la DMD/DMB. Se ha demostrado que el uso de glucocorticoides ralentiza la progresión de la escoliosis y la pérdida de la deambulación 26. Respecto a la función motora, se ha sugerido que mejoran la fuerza muscular y las pruebas de función cronometradas estandarizadas (27, 28). Algunos estudios sugieren que su uso mejoraría la supervivencia y retrasaría la aparición de la miocardiopatía 26. Debido a estos beneficios el uso de esta terapia es una necesidad en estos pacientes. Sin embargo, en el uso prolongado es importante lograr un mayor beneficio posible. Ante ello, es necesario evaluar la eficacia y seguridad del uso de los glucocorticoides.
De la evidencia a la decisión:
Seleccionamos tres RSs que reportan mayor fuerza muscular para el grupo tratado con prednisona a dosis de 0,75/kg/día comparado con el placebo; siendo la fuerza muscular medida con la escala desarrollada por el Consejo de Investigación Médica de Reino Unido (MRC, por sus siglas en inglés), tiempo de marcha de 9 metros, tiempo de marcha de 10 metros y tiempo para subir cuatro escaleras. De la misma manera, los desenlaces de funcionalidad, que incluyen la capacidad vital forzada (CVF), y de calidad de vida se mostraron favorables para la prednisona. Los principales eventos adversos (EA)fueron el cambio de peso corporal y los cambios de comportamiento, encontrando una diferencia de medias (DM) de 1.74 (IC95 % -0.33 a 3.81) y un RR 1.39 (IC95 % 0.94 a 2.06) mayor en el grupo con prednisona, respectivamente. Además, al comparar los efectos de la prednisona diaria con una vez cada fin de semana, los resultados fueron a favor de prednisona cada fin de semana. Finalmente, al medir los efectos de prednisona 0,75/kg/día contra deflazacort 2 mg/kg/día, los desenlaces de fuerza muscular y de seguridad se mostraron a favor del deflazacort.
El conjunto de evidencia presentó elevado riesgo de sesgo y una imprecisión elevada, resultando en una certeza global muy baja a moderada. Sin embargo, por ser aceptable, factible y aplicable el GEG formuló una recomendación fuerte a favor del uso de glucocorticoides en el tratamiento de DMD.

Pregunta 5: En pacientes con DMD con mutaciones sin sentido, ¿es eficaz y seguro el tratamiento con ataluren más corticoesteroides comparado con solo corticoesteroides?
Ataluren es un medicamento aprobado en varios países, incluyendo todos aquellos asociados a la Unión Europea, para el tratamiento de mutaciones sin sentido en pacientes con DMD
Para cada desenlace crítico o importante de cada pregunta PICO, se buscó RS(publicadas como artículos científicos o como parte de una GPC) (DMDss) 29. Es administrada por vía oral. Su mecanismo de acción se basa en la promoción de la lectura ribosomal, evitando la detención prematura de traducción genética. De esta forma, permite producción de la proteína de distrofina (30, 31). EL GEG consideró importante determinar su utilidad en términos de eficacia y seguridad es de importancia para que posteriormente se puedan establecer estrategias de financiamiento para los pacientes que la requieran.
De la evidencia a la decisión:
En 2 estudios seleccionados, se reportó para ataluren una mayor distancia recorrida en la prueba funcional de 6 minutos, con DM de 17.2 metros más (IC95 %: 0.2-34.1). En la prueba funcional de 10 metros, una DM de 1.1 segundos menos (IC95 %: 2.2-0.1) en el tiempo de caminata. En las pruebas de pronóstico, que incluyeron el riesgo de empeoramiento del 10 % en la caminata de 6 minutos y pérdida de deambulación, fueron favorables para Ataluren con HR de 0.68 (IC95 %: 0.48 a 0.94) y 0.28 (IC95 %. 0.19-0.42) respectivamente. Los EA reportaron una frecuencia de 45.6 % y 33.9 % de eventos adversos atribuibles al Ataluren, sin embargo, ninguno de ellos fue severo.
El conjunto de evidencia, con imprecisión elevada, presentó certeza global moderada. Aunque, su principal barrera es su alto costo, tiene un balance a favor del beneficio, es aceptable y aplicable, por lo cual el GEG formuló una recomendación condicional a favor del uso de ataluren en el tratamiento de DMD.

Pregunta 6: En pacientes con Distrofia Muscular De Duchenne susceptibles de omitir el exón 51 ¿es eficaz y seguro el tratamiento con eteplirsen más corticoesteroides comparado con corticoesteroides?

Eteplirsen es un medicamento aprobado en la Unión Europea y Estados Unidos usado para el tratamiento de DMD 32. Es administrado por vía endovenosa. Los oligonucleótidos sintéticos antisentido, en pacientes con determinadas deleciones, candidatas a esta intervención, permiten la omisión del exón objetivo, salto de exón, o “exon skkiping” en inglés, alrededor de la deleción heredada en el pre-ARN mensajero y restaura el marco de lectura; el eteplirsen se une al exón 51 del pre-ARN mensajero, provoca la omisión de este exón en el transcripto final y restaura el marco de lectura, aumentando así la producción de distrofina, truncada pero funcional 33. El GEG consideró importante determinar su utilidad para los pacientes que la requieran.
De la evidencia a la decisión:
Se seleccionaron 5 estudios que mostraron resultados funcionales favorables para el uso eteplirsen con una diferencia media de 67.3 m (27.32 - 107.28) en la distancia de caminata de 6-minutos, un mayor porcentaje de expresión de distrofinas y un ligero incremento anual de 3.37 % (1.93 - 4.8) de la CVF. Los EA fueron principalmente respiratorios (25 %) y procedimentales (20 %), siendo leves.
El conjunto de evidencia mostró, con alto riesgo de sesgo, inconsistencia e imprecisión, certeza global muy baja. Aunque su principal barrera es su alto costo, presentó un balance a favor del beneficio, es aceptable y aplicable, por lo cual el GEG formuló una recomendación condicional del uso de eteplirsen en el tratamiento de DMD.

Pregunta 7: En pacientes con DMD susceptibles de omitir el exón 53, ¿es eficaz y seguro el tratamiento con Golodirsen más Corticoesteroides comparado con Corticoesteroides?
El golodirsen (Exón 53 PMO, SRP 4053 o Vyondys 53) es un oligonucleótido antisentido de la subclase oligómero de morfolino fosforodiamidato (PMO) diseñado para omitir específicamente el exón 53. Los pacientes con mutaciones del gen de distrofina susceptibles a la omisión de dicho exón representan aproximadamente el 8-10 % de todos pacientes con DMD (34-36). Hasta el momento de la elaboración de la GPC, un ensayo fase I/II ha reportado que estas terapias aumentan la expresión de distrofina en laboratorio pero no se ha demostrado aún beneficios clínico 37. La omisión de exones como terapias génicas parece ser una opción beneficiosa en pacientes con DMD susceptibles a la omisión del exón 53 aunque sus resultados aún son primarios biológicos 38. Su aplicabilidad puede ser un desafío, sobre todo, en países de bajos ingresos. El GEG consideró importante determinar su utilidad para los pacientes con DMD susceptibles.
De la evidencia a la decisión:
Se seleccionó un estudio que reportó una media de aumento de 16 veces de la expresión de distrofina, una media de aumento de 13.46 veces (DE=11.91, rango: 1.88 a 49.67) (p < 0.001) en el porcentaje de fibras musculares positivas para distrofina y un aumento de 28.89 veces (DE=39.67, rango de 2.59 % a 150.36 %) en el porcentaje de omisión del exón 53. Sin resultados en desenlaces funcionales. Todos los pacientes presentaron al menos un efecto adverso leve.
El conjunto de evidencia presentó, con alto riesgo de sesgo, imprecisión y evidencia indirecta, certeza global muy baja a baja. Aunque su principal barrera es su alto costo, presenta beneficios biológicos, es aceptables y aplicable, por lo cual el GEG formuló una recomendación condicional del uso de golodirsen en el tratamiento de DMD.

Pregunta 8: En pacientes con DMD susceptibles de omitir el exón 53, ¿es eficaz y seguro el tratamiento con Vitolarsen más Corticoesteroides comparado con Corticoesteroides?
El viltolarsen (NCNP-01; NS 065; NS-065 / NCNP-01 o VILTEPSO®) es un oligonucleótido antisentido de la subclase oligómero de morfolino fosforodiamidato (PMO) diseñado para omitir específicamente el exón 53. A diferencia del golodirsen, el viltolarsen es un oligonucleótido de 21 nucleótidos, mientras que golodirsen posee 25 39. Su uso ha sido aprobado en USA y Canadá 40. Sin embargo, su aplicabilidad puede ser un desafío, sobre todo, en países de bajos ingresos. El GEG consideró importante determinar su utilidad para los pacientes con DMD susceptibles.
De la evidencia a la decisión:
Se seleccionaron 3 estudios que mostraron un incremento en el nivel de la expresión de distrofina de 5,7 % (DE:2.4 %) en la cohorte de 40mg/kg y del 5,9 % (DE 4.5 %) en la cohorte de 80 mg/kg, sin declarar una diferencia estadísticamente significativa. De la misma manera, hubo un aumento en el porcentaje de miofibras positivas para distrofina de 14.3 % (DE 7.8 %) en el grupo de 40mg/kg y 34.8 % (DE 20.4 %) en el grupo de 80mg/ kg. Con respecto al nivel de omisión del exón 53 se reportó un aumento para la dosis de 40 mg/kg (de 17.4 % a 28 %) y para 80mg/kg (de 43.9 % a 49.7 %). Los desenlaces funcionales, medidos con la prueba de caminata de 6 minutos y el tiempo para caminata/correr de 10 metros, fueron favorables para el vitolarsen, sin embargo, no reportaron una significancia estadística. Finalmente, 80-100 % de participantes presentaron EA leves.
El conjunto de evidencia presentó, con alto riesgo de sesgo, imprecisión y evidencia indirecta, certeza global muy baja a baja. Aunque su principal barrera es su alto costo, presenta beneficios biológicos, es aceptables y aplicable por lo cual el GEG formuló una recomendación condicional del uso de golodirsen en el tratamiento de DMD.

Agradecimientos
Agradecimientos: A la Sociedad Peruana de Neurología, por el financiamiento de la presente guía.
Representantes de pacientes: María Rodríguez Berckermeyer, Federación Peruana de Enfermedades Raras (FEPER); Luis Miguel Del Águila, Georgina Guerrero Torres, Anel Townsend Diez-Canseco, Maribel Bejarano Massolo, Asociación de Distrofia Muscular del Perú (ADM-Perú); Rafael Zegarra León, Duchenne Parent Project Perú; Alexandra Grau Quinteros, Fundación Ruedas Mágicas.
Revisores Metodológicos: Dra. Isabel Pinedo Torres. Médico especialista en Endocrinología del Hospital Nacional Daniel Alcides Carrión. Master en Epidemiología Clínica. Docente investigador de la Universidad Científica del Sur. Dr. Wilfor Aguirre Quispe. Master en Epidemiología Clínica. Past - Médico evaluador y supervisor de ensayos clínicos de la Oficina General de Investigación y Transferencia Tecnológica (OGITT) del Instituto Nacional de Salud (INS) del MINSA - Perú. Malaga Julca Marco Moises, Huerta Rosario Andrely Cristina, Pacheco Barrios Niels y Rodriguez Calienes Aaron, miembros de la Red de Eficacia Clínica y Sanitaria (REDECS)
Revisores Externos: Dr. Alberto L. Dubrovsky. Profesor Titular de Neurociencias - Universidad Favaloro. Profesor Adjunto de Neurologia - Universidad de Buenos Aires. Director del Departamento de Neurología y Unidad de Enfermedades Neuromusculares. Instituto de Neurociencias Fundación Favaloro. Dr. Carlos Ignacio Ortez Gonzáles. Médico Neurólogo Pediatra, especialista en enfermedades neuromusculares, Hospital Sant Joan de Déu , Barcelona España.

Grupo de validación.
Ricardo Fujita Alarcón, Centro de Genética y Biología Molecular, Facultad de Medicina Universidad de San Martín de Porres, Lima, Perú.
Aníbal Prentice De Lama, Clínica Angloamericana, Lima, Perú. Mario Cornejo Olivas, Instituto Nacional de Ciencias Neurológicas, Lima, Perú.
Pilar Medina Alva, Instituto Nacional Materno Perinatal, Lima, Perú. Hugo Abarca Barriga, Instituto Nacional de Salud del Niño, Lima, Perú.

