










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de la Sociedad Química del Perú
Print version ISSN 1810-634X
Rev. Soc. Quím. Perú vol.71 no.1 Lima Jan./mar. 2005
Amiduros de los metales del Grupo del Platino
Gabriel García-Herbosa1
1 Área de Química Inorgánica. Departamento de Química. Facultad de Ciencias. Universidad de Burgos. España E-mail: gherbosa@ubu.es
Los compuestos en los que está presente, al menos, un enlace metal-nitrógeno del tipo R 2 N-M, resultante de sustituir un átomo de hidrógeno en una amina por un metal, se conocen como amiduros, amidas o amidocomplejos.
El objetivo de esta exposición es hacer una presentación de la química de amiduros de los metales del grupo del platino. Se trata de compuestos de gran importancia por el papel que desempeñan como intermedios en reacciones de formación de enlaces C-N, tales como la hidroaminación de olefinas o la síntesis de arilaminas a partir de haluros de arilo. Otras interesantes reacciones en las que intervienen los complejos amido, como la hidrogenación de iminas, se co-mentarán durante esta exposición.
Los amiduros guardan relación isoelectrónica con los s-alquilos (R 3 C-M) y los alcóxidos (RO-M) y por ello exhiben estequiometrías, estructuras y volatilidades similares. Con frecuencia muestran aspectos comunes, como las reacciones de b-eliminación características de los s-alquilos, la formación de puentes, que es típica de los alcóxidos, y las reacciones de inserción y de eliminación reductora que comentaremos a lo largo de la sesión. Pero la abundancia de complejos s-alquilo contrasta con la relativa escasez de amido complejos de metales del grupo del platino, lo que puede interpretarse dado el carácter blando de estos metales y duro de los ligandos amido. Es precisamente esa relación ácido blando/base dura la responsable de la interesante reactividad que muestran los complejos amido.
En la última parte de la sesión y sobre la base del conocimiento actualmente existente sobre las reacciones que conectan complejos amina (M-NHR-CHR 2 ) con complejos imina (M-NR=CR 2 ), que difieren en una molécula de hidrógeno, se presentará una propuesta de un ciclo catalítico para la descomposición fotoquímica de agua que podría tener futuro en el ámbito de la conversión y el almacenamiento de la energía solar.
INTRODUCCIÓN
En primer lugar quiero agradecer al Comité Organizador del vigésimo segundo Congreso Peruano de Química y en especial al profesor Jorge Angulo la amabilidad que ha tenido al invitarme a impartir esta charla. Me gustaría también aprovechar la ocasión para recordar que no es posible entender el progreso sin una constante dedicación a la investigación, sin un constante esfuerzo de los gobiernos para apoyar y fortalecer la investigación. Y ese esfuerzo también ha de venir de quienes nos dedicamos a esta bonita tarea y aprovecho la ocasión para animar a todos los investigadores y especialmente a los más jóvenes a entusiasmarse con la investigación, a pesar de las limitaciones y obstáculos que podamos encontrar cada uno en nuestro ámbito. Sólo a través de la investigación en los diferentes campos de la ciencia puede alcanzarse el bienestar y el progreso de la sociedad.
Les voy a hablar de un tipo de compuestos químicos que denominamos amiduros y también complejos amido. Para ello mi intervención se organiza como vemos en el índice. En primer lugar una descripción del enlace M-N amido. Después comentaré algunas reacciones de preparación de estos compuestos. Continuaremos analizando en qué radica la importancia de estos compuestos, mencionando especialmente las reacciones de formación de enlaces C-N de gran importancia en química. Después les mostraré algunos resultados alcanzados en nuestro laboratorio y finalmente un proyección hacia el futuro, hacia cómo podrían los complejos amido ayudar en un asunto tan importante como es la conversión y el almacenamiento de la energía solar en forma de hidrógeno.

Mi llegada a este tipo de compuestos se produjo, como tantas veces ocurre en investigación, por azar, con la síntesis del complejo que se muestra y que nos sirve para iniciar la introducción al enlace M-N amido porque es importante diferenciar ese enlace covalente del enlace coordinado entre un metal de transición y el nitrógeno.
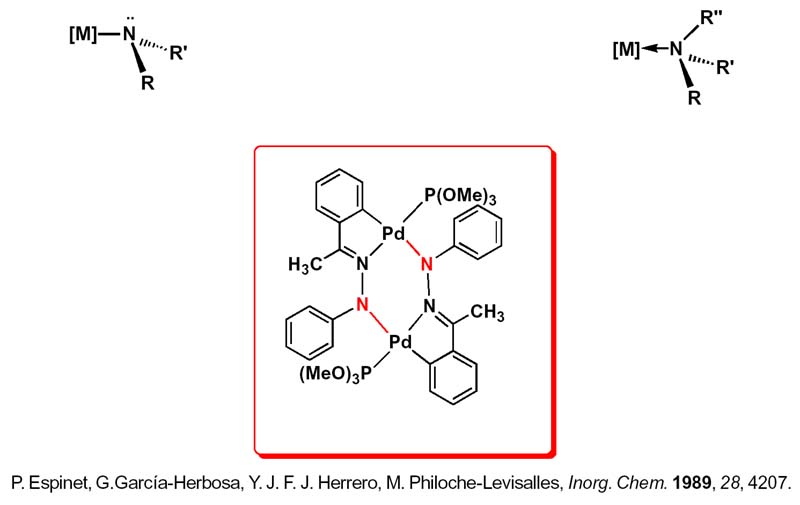
A los compuestos con enlace metal-nitrógeno R 2 N-M, resultantes de cambiar un átomo de hidrógeno en una amina por un metal, los denominamos de tres posibles modos: amiduros, amidas o amidocomplejos y están íntimamente relacionados con los alquilos R 3 C-M y los alcóxidos RO-M con los que guarda relación isoelectrónica y manifiestan estequiometrías, estructuras y volatilidades similares -ver los de Cr(IV)- y otras propiedades comunes como por ejemplo la ß-eliminación característica de los s-alquilos y la formación de puentes típica de los alcóxidos, reacciones de inserción, y de eliminación reductora que tendremos ocasión de comentar después.

Aunque como indica el título de la charla nos vamos a centrar en amidocomplejos de algunos metales del grupo del platino es conveniente, en mi opinión, hacer un breve repaso de este tipo de compuestos siguiendo la tabla periódica para reconocer las diferencias entre los amiduros en función de la naturaleza del metal al que se enlazan.
Los amidocomplejos de los metales alcalinos , en especial los de Li, son reactivos muy importantes en química orgánica y organometálica, con estructuras moleculares dímeras, trímeras e incluso tetrámeras en las que el ligando amido actúa como puente. Se trata de buenos compuestos de partida para la síntesis de amidocomplejos de otros metales.
La volatilidad de estos amiduros los hace interesantes en aplicaciones diversas, como por ejemplo, puede usarse como fuentes dopantes la CVD (chemical vapor depossition) de GaN:Mg. [Jr., 2000 #1].
Los amiduros del grupo 13 tienen también estructuras con amiduro puente, aunque con sustituyentes voluminosos se conocen monómeros. Los del grupo 14 tienden a ser dímeros o monómeros con ligandos voluminosos.
Los amiduros de los metales de los grupos representativos, además de las características estructurales comunes, se muestran especialmente sensibles a la humedad dando la reacción de hidrólisis esperada, liberando la amina y el hidróxido o el óxido metálico hidratado.
Los amidocomplejos de los metales de transición muestran un comportamiento variable, dependiendo del carácter duro o blando del centro metálico. Con centros metálicos duros como Ti(IV), Zr(IV) y Hf(IV) y V(V) , Nb(V) Ta(V) e incluso lantánidos y actínidos, podemos encontrar compuestos mononucleares como Ti(NR 2 ) 4 (análogo al de Cr(IV) que utilizábamos como ejemplo al principio y que experimentan reacción de hidrólisis análogamente a los alcóxidos; más difícil cuanto más larga es la cadena hidrocarbonada y el grado de condensación o polimerización.
Especial mención merece el compuesto de Mo(III) con el ligando bis-2,6 diisopropilfenilamiduro, (Laplaza 1995) muy sensible a la humedad, que reacciona con nitrógeno a temperatura ambiente rompiendo el triple enlace de la molécula de nitrógeno y dando un nitruro que se puede transformar fácilmente en amoniaco. El interés de estos amidocomplejos es natural por la relación evidente que guardan con las molibdoenzimas nitrogenasa.
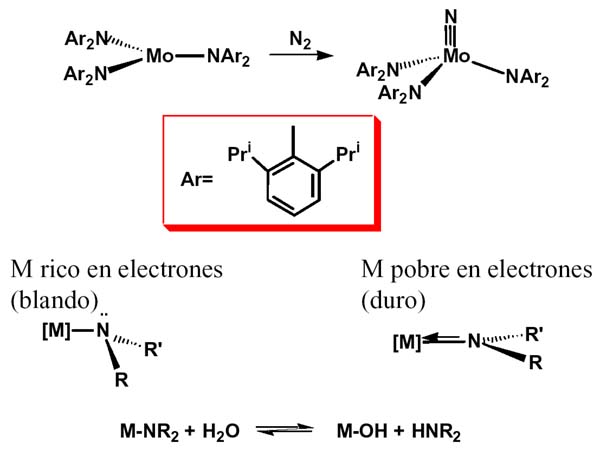
Pero a medida que nos desplazamos a la derecha en la tabla periódica, hacia metales ricos en electrones, comienzan a cambiar las cosas y se produce un vacío considerable en cuanto a ejemplos bien caracterizados, aunque en los últimos años es evidente que los esfuerzos dirigidos a su síntesis están dando resultados y su número aumente paulatinamente. Para comprender la diferencia podemos fijarnos en la estructura del enlace amido.
Cuando el ligando amido es terminal podemos esperar estructuras piramidal y plana, dependiendo de que el par de electrones se implique o no en enlacep. Ello dependerá, en buena medida, de la riqueza electrónica del metal. Si el metal es un centro duro, con elevada carga, la participación p es previsible y así la mayoría de los amidocomplejos que hemos mencionado hasta ahora y, en general la mayoría de los que se conocen, son planos. Además, en la medida que el par solitario se implique en enlacep, la formación de puentes estará menos favorecida que si el par electrónico está disponible.
La menor abundancia de amidocomplejos de metales del grupo del platino está relacionada, muy probablemente, con la relativa riqueza electrónica de estos metales de la derecha de las series de transición o, en otras palabras, del contraste entre el carácter blando del metal y duro del ligando.
Hay que destacar que la reacción de hidrólisis que hemos mencionado puede invertirse cuando se trata de un metal del grupo del platino, lo que indica una diferencia notable de comportamiento entre unos amidocomplejos y otros.
En cuanto a los métodos de preparación de complejos amido hay que señalar que no existe un método general. Los relativamente escasos ejemplos de amidocomplejos de metales de transición contrasta con el elevado número de métodos de preparación. Podemos decir que cada caso ha sido preparado mediante una reacción diferente. Todo parece indicar que si termodinámicamente no son muy favorables, llegar a ellos exige a veces rutas cinéticas extrañas que no permitan llegar a otros compuestos termodinámicamente más favorables.
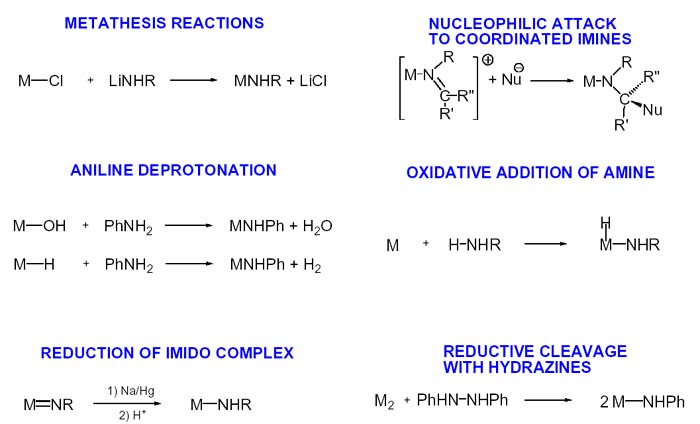
La reacción de metátesis es muy lógica y conducida por la formación de LiCl.
La desprotonación de anilina por un complejo hidroxo (esta es el inverso de la hidrólisis que hemos comentado), o por un hidruro
dando hidrógeno. Llamo la atención sobre esta reacción que puede ser de utilidad a los efectos de lo que contaré en la última parte de la intervención. El ataque nucleofílico a una imina, que se publicó como probablemente generalizable y luego no resultó serlo (ninguna de las que se muestran lo es).
La adición oxidante de una amina es también una reacción de enorme interés (recordemos que se produce un aumento en dos unidades del estado de oxidación del metal) y podemos utilizarla para diseñar un ciclo catalítico para la conversión y almacenamiento de energía solar mediante activación (ruptura) de la molécula de agua por complejos amido.
A pesar de la relación isoelectrónica con los s-alquilos, el enlace M-C propio de los compuestos organometálicos, es ciertamente extraño en los sistemas biológicos; mientras que el enlace amido lo utiliza la naturaleza en muchos compuestos capaces de llevar a cabo algunas de las reacciones más importantes para la existencia de la vida, como las de transferencia de electrones en los citocromos, transporte de oxígeno en hemoproteínas, reacciones fotoquímicas rédox en las clorofilas, o en la superoxidodismutasa de Cu y Zn a través del imidazolato puente. En la coenzima B12, un producto natural extraordinario en muchos aspectos, también lo es en el de presentar un enlace Co-C en cis a un enlace Co-N amido, disposición favorable a experimentar eliminación reductora con formación de enlace C-N, reacción que evidentemente no ocurre.
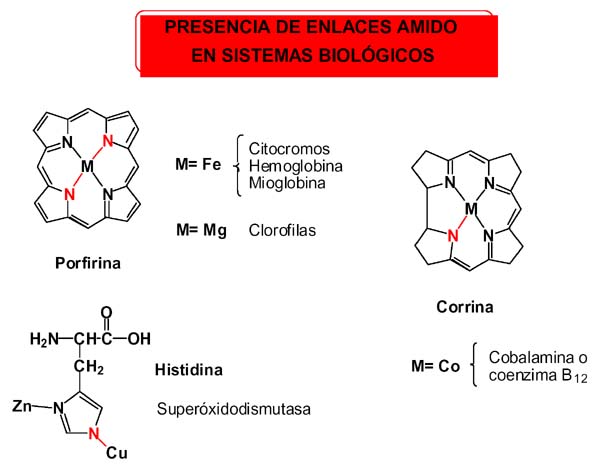
Hay que hacer notar que en todos los compuestos de origen biológico el enlace amido está conjugado entre los extremos de descripción del enlace amido e imino, lo que debe ayudar a su estabilización.
¿Qué es lo que mantiene el interés por los amidocomplejos, en general, y de los metales del grupo del platino en particular?.
Sin duda, el interés principal por los amidocomplejos de los metales de transición se centra en el papel que realizan en importantes reacciones de formación de enlaces C-N en las que los amidocomplejos se postulan como intermedios catalizadores.
Las aminas forman una clase de compuestos de gran importancia tanto como productos finales, como intermedios versátiles en procesos de interés industrial.
Tres aproximaciones importantes a la síntesis de aminas voy a mencionar, la hidroaminación de alquenos, la síntesis de arilaminas a partir de haluros de arilo y la hidrogenación de iminas.
Probablemente la reacción de mayor interés es la hidroaminación de olefinas [Arredondo, 1999; deBruin, 2000; Roesky, 1997 ; Roundhill, 1997 ; Senn, 2000; Shaver, 2000; Uchimaru, 1999]. La reacción de hidroaminación de olefinas es factible desde un punto de vista termodinámico, siendo exoérgica y exotérmica en condiciones normales. Sin embargo, la entropía de reacción es negativa, lo que impide el uso de elevadas temperaturas para vencer la barrera de activación necesaria para la reacción de adición directa a temperatura ambiente. Por ello se requieren catalizadores que abran pasos de baja energía. La reacción catalizada puede transcurrir por dos rutas, la de activación del alqueno y de la de activación del enlace N-H.
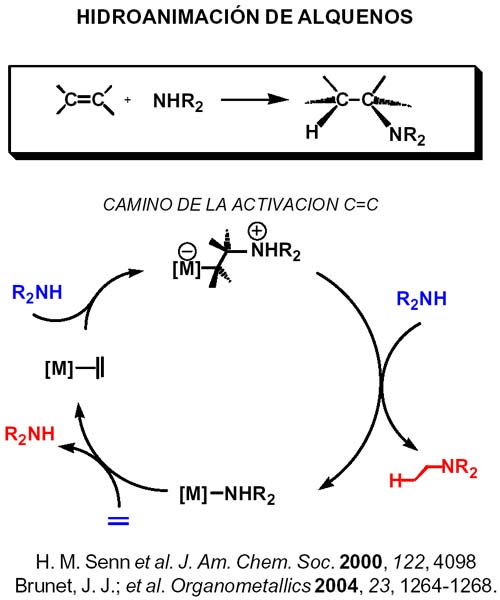
En la ruta a través de la activación del alqueno, cada una de cuyas etapas ha sido sometida a cálculos basados en la teoría de la densidad funcional en la referencia que aparece debajo, el doble enlace, está activado por coordinación, y el enlace C-N se forma por ataque nucleófilo de la amina al alqueno coordinado.
Para liberar el producto hay que escindir o romper el enlace M-C del complejo alquilo, lo que puede hacerse mediante protonolisis directa en la que el protón se inserta en el enlace M-C a través de un intermedio de tres centros metal-hidrógeno-carbono (favorecida en grupo 10, Ni, Pd, Pt según los cálculos mencionados), o por protonación del centro metálico seguida de eliminación reductora(favorecido en grupo 9 Co, Rh, Ir) del producto quedando el aminocomplejo del que se desplaza la amina por interacción con el alqueno.
En la referencia que se cita se reproducen las siguientes conclusiones:
1. El ataque nucleófilo de la amina al alqueno es termodinámica y cinéticamente favorable para los metales del grupo 10, pero muy desfavorecido para el grupo 9 donde constituye la etapa limitante.
2. La etapa de escisión del enlace M-C es la etapa limitante en el grupo 10 siendo fácil para el grupo 9.
3. En promedio, los metales del grupo 10 parecen más adecuados (el Ni el más prometedor).
4. La ß-eliminación en el alquilamoniocomplejo es una reacción colateral competitiva cinéticamente, pero desfavorecida termodinámicamente en la ruptura protonolítica.
La etapa clave para los metales del grupo 10 es, como hemos dicho, la ruptura del enlace M-C y la protonolisis parece la ruta más conveniente y puede tener lugar como se muestra en la transparencia siguiente. (En 2004 se publica por vez primera la hidroaminación de etileno con anilina catalizada por complejos de platino. (Brunet, J. J.; Cadena, M.; Chu, N. C.; Diallo, O.; Jacob, K.; Mothes, E. Organometallics 2004, 23, 1264-1268.)
La protonolisis directa puede llevarse a cabo por varias vías 1) el propio ion amonio adoptando una disposición favorable a la formación del puente metal-hidrógeno- carbono 2) a través de otra amina que se encarga de aportar un protón al enlace M-C, lo que puede hacerse con o sin coordinación al centro metálico(vía enlace de hidrógeno amina-amonio), la diferencia está en que si se realiza con coordinación lo hace a través de un amido-intermedio que reacciona con el alquilamonio liberado proporcionando la amina y el aminocomplejo. El aminocomplejo reacciona con el alqueno continuando el ciclo catalítico.
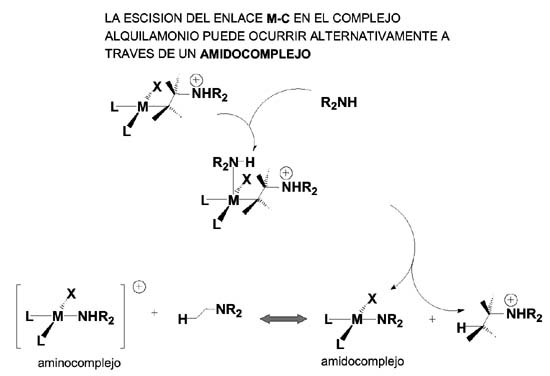
Otra posible ruta para la hidroaminación de alquenos es la denominada de activación N-H y, aunque se ha anunciado la publicación de cálculos del mismo tipo que para la ruta de la activación del alqueno que aún no han salido a la luz, de nuevo nos podemos encontrar con dos alternativas:
a) La activación mediante adición oxidante del enlace N-H, dando el complejo hidruro-amiduro. Después la insercción del alqueno que bien puede tener lugar en el enlace M-H (conduciendo a un complejo cis alquilo-amido) o en el enlace M-N amido. En cualquiera de los casos, la siguiente etapa de eliminación reductora conduce a la amina y libera el catalizador. El ejemplo más representativo de este ciclo es el descrito por Casalnuovo[Casalnuovo, 1988] basado en Ir(I) para hidroaminación de norborneno con anilina aunque con muy baja actividad. También utilizando iridio se ha publicado la primera hidroaminación enantioselectiva[Dorta, 1997] y más recientemente aún complejos de lantánidos[Molander, 1999] también se muestran útiles en hidroaminación enantioselectiva.
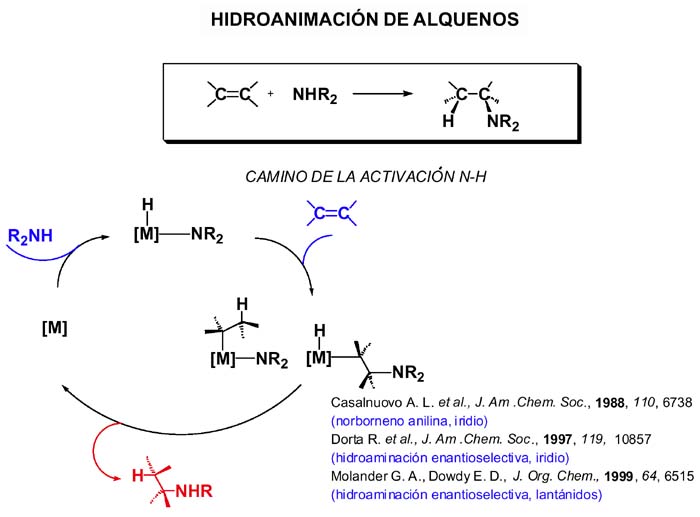
b) En lugar de activarse por adición oxidante el enlace N-H puede hacerlo por desprotonación dando el amiduro que inserta el alqueno. El ciclo catalítico que se propone a continuación permite la hidroaminación del alqueno partiendo de un amidocomplejo y aceptando los siguientes supuestos: El catalizador se genera in situ por ruptura del puente amido (situación bien conocida la de formación de puentes). La amina al coordinarse desprotona; eso es razonable pues se vuelve más ácido el enlace N-H protonándose el amiduro inicial que pasa a amina. Eso requiere que el protón viaje de la amina coordinada a la posición amido inicial. La diferente acidez será crítica para asegurar el éxito del ciclo. Después la inserción del alqueno en el enlace amido y, finalmente, el protón ha de transferirse desde el nitrógeno amino al alquilo que se libera.
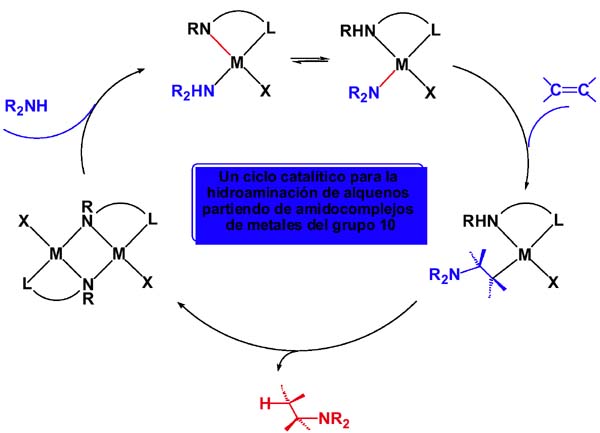
Como veremos después, podemos obtener complejos binucleares de paladio con amiduros puente de forma diastereoespecífica, y eso puede ser interesante desde el punto de vista de la catálisis pues tal vez se induzca selectividad en la disposición geométrica de los grupos en torno al enlace C-N de la amina que se libera.
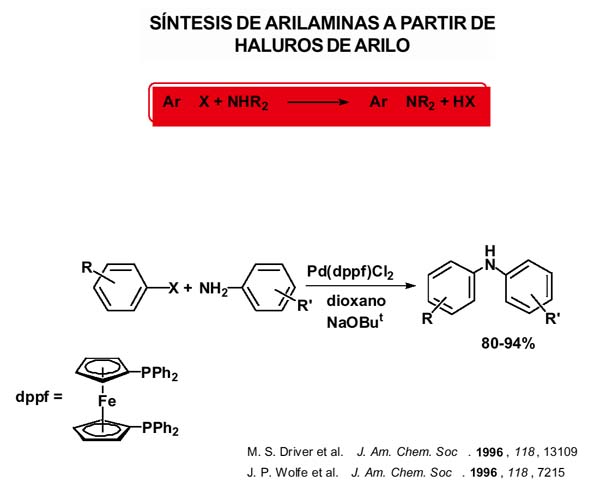
La segunda reacción que vamos a considerar es la síntesis de arilaminas a partir de haluros de arilo [Hartwig, 1998]. A pesar de la aparente simplicidad de las arilaminas, su síntesis es a menudo muy difícil. La nitración, reducción o sustitución son a menudo incompatibles con muchos otros grupos funcionales y requieren etapas de protección y desprotección. La sustitución nucleofílica directa de haluros de arilo requiere, en general, gran exceso de reactivo, un disolvente de elevada polaridad y, o elevadas temperaturas o haluros muy activados.
En 1996 se publica la reacción de aminación de diversos bromuros y yoduros de arilo catalizada mediante complejos de paladio con ligandos fosfina lábiles como el que se muestra con un grupo metilo en posición orto que introduce notable impedimento estéreo en presencia de bases (alcóxidos o sililamidas). Los mejores sustratos son las aminas secundarias. Sin embargo, cuando el complejo de paladio contiene el ligando difenilfosfinaferroceno se obtienen rendimientos muy elevados con alquilaminas primarias, secundarias cíclicas y anilinas, como el ejemplo que se muestra para preparar difenilaminas sustituídas. Algunos ejemplos de aminación intramolecular también aparecen en 1996. Resulta irónico pensar que inicialmente se consiguiera aminación con ligandos lábiles fosfina y que se consiguiera una enorme mejora usando una fosfina quelante.
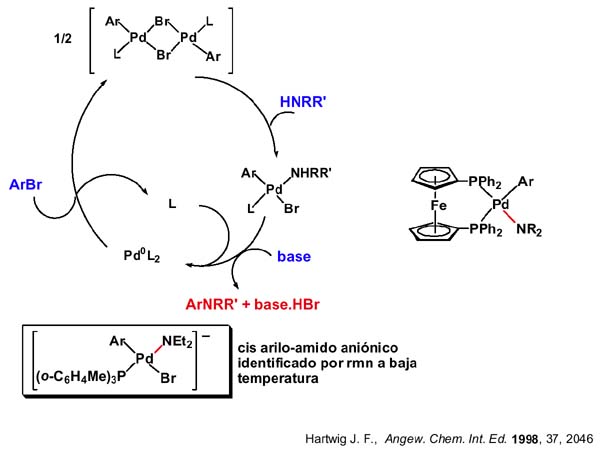
El mecanismo que se propone es la eliminación reductora del cis-arilo-amido complejo y la consecutiva adición oxidante del haluro de alquilo al complejo de paladio(o). En la transparencia se muestra un ejemplo de un ciclo bien caracterizado en el que L es la triortotolilfosfina. El aril complejo dímero de bromos puente reacciona con la amina y la reacción con base conduce al amidocomplejo que en este caso pudo ser identificado por resonacia magnética nuclear a baja temperatura. El complejo de paladio cero resultante de la eliminación reductora experimenta adición oxidante del bromuro de arilo cerrando el ciclo.
La eliminación reductora C-N es la etapa clave en estas aminaciones catalizadas. Los ejemplos de eliminaciones reductoras C-N son escasos comparados con las eliminaciones reductoras C-C y C-H lo cual es lógico si consideramos la escasez de amidocomplejos comparada con hidruros y alquilos. En general, la química organometálica catalítica que conduce a enlaces carbono-heteroátomo es menos conocida y está menos desarrollada que la de la formación de enlaces C-C (el mecanismo detallado por el que ocurre el proceso Wacker de oxidación de etileno a acetaldehido sigue sin comprenderse muy bien y la oxidación de hidrocarburos continúa siendo área de investigación activa ).
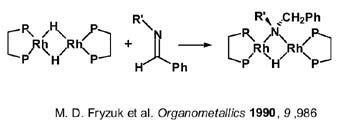
Ejemplos de compuestos bien caracterizados que experimentan estas reacciones, sólo se conocen desde 1996. Actualmente hay varios ejemplos y los factores que controlan esa reacción comienzan a comprenderse. El mecanismo de la eliminación reductora considera que los enlaces M-N amido y M-C han de estar en posición cis; sin embargo, los ejemplos bien caracterizados se limitan a los del tipo bisfosfinaferroceno, de reciente preparación, en los que se observa eliminación reductora más fácil por este orden alquilamido>arylamido>diarylamido, es decir, cuanto más nucleófilo el amido, más rápida la reacción de ER, y cuanto más electrófilo es el grupo arilo también resulta más fácil la ER.
ß-eliminación. En los casos estudiados con alquilamido, la reacción competitiva de ß-eliminación ha resultado más lenta (porque no se ha producido) que la eliminación reductora de C-N. Aunque la ß-eliminación en amidocomplejos de metales de transición de la parte derecha, ricos en electrones, se presumía rápida no se encuentran casos de ß-eliminación irreversibles hasta muy recientemente. De hecho, Fryzuk describe un sistema reversible que se muestra en la transparencia que sugiere que el proceso de inserción y el inverso de eliminación son térmicamente neutros o su balance térmico es próximo a cero.
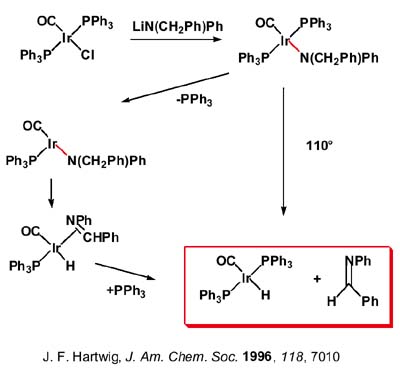
El amidocomplejo de Ir, derivado del compuesto de Vaska, que se muestra arriba a la derecha, es uno de los ejemplos que han surgido en los últimos años basado en la presencia de fosfinas en trans que impiden la formación de puentes y estabilizan el complejo como ligandos p-aceptores que retiran la carga que introduce el ligando amido. El complejo experimenta ß-eliminación pero requiere elevada temperatura. El mecanismo de la ß-eliminación implica amidocomplejos monómeros, que disocian a un complejo intermedio tricoordinado (aunque conclusiones firmes sobre el número de coordinación del intermedio requieren estudios más detenidos porque un intermedio pentacoordinado tampoco debe ser descartado) que sufre ß-eliminación de modo análogo a los sigma-alquilos pasa por el iminocomplejo que, finalmente, libera la imina no habiéndose detectado el iminocomplejo en sistemas con ligandos amido monodentados. En nuestro laboratorio, como veremos después, hemos encontrado complejos imino procedentes de la ß-eliminación usando ligandos bidentados quelatos y hemos caracterizado complejos que retienen la imina que se libera en el proceso de ß-eliminación.
En resumen, podemos afirmar que el conocimiento de los factores que gobiernan la eliminación reductora y ß-eliminación son claves para encontrar los catalizadores adecuados para conducir el proceso de aminación lo más eficazmente posible.
En tercer lugar, vamos a comentar la presencia de los amido complejos en la reacción de hidrogenación de iminas catalizada, como ruta de síntesis de aminas, especialmente aquéllas quirales, cuya importancia demanda sistemas catalíticos eficaces para la reducción asimétrica de iminas por parte de las industrias farmaceúticas y agroquímicas.
Aunque las bases de la activación de dihidrógeno mediante metales de transición están bien establecidas, el desarrollo en la comprensión de la hidrogenación catalizada de dobles enlaces C=X(heteroátomo)decrece de olefinas a cetonas y a iminas decrece por este orden y es relativamente escaso el conocimiento sobre activación de iminas y las requeridas etapas de transferencia de átomos de hidrógeno.
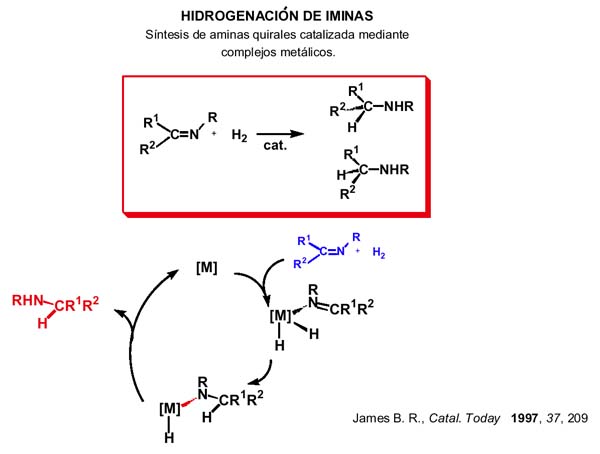
Desde un punto de vista termodinámico, la hidrogenación de iminas es ligeramente exotérmica(-60kJmol-1) la de alquenos es del orden de –130kJmol -1 lo que explica, en parte, la mayor dificultad de hidrogenar iminas comparado con la de alquenos. Por otro lado, el enlace side-on de las olefinas que proporciona un buen solapamiento orbital, lo que se considera un requisito importante en el ciclo catalítico de la hidrogenación, no está bien establecido en metales del grupo del platino donde el enlace es típicamente end-on a través del nitrógeno imina.
La hidrogenación puede realizarse directamente con hidrógeno o bien mediante un agente de transferencia de hidrógeno o dador de hidrógeno en presencia de catalizadores. La clave del asunto está en la discriminación que puede realizar un complejo metálico quiral de las dos enantiocaras de una imina en el nitrógeno sp 2 en la etapa de la transferencia de protón que se incorpora al N-amido produciendo reducción asimétrica de iminas con elevados (>90%) ex-cesos enantioméricos. Como veremos más adelante, nos hemos encontrado con varias experiencias de deshidrogenación de aminas coordinadas (el camino in-verso) que puede ayudar a entender el mecanismo de la hidrogenación/ deshidrogenación y los factores implicados.
Brian R. James ha revisado la hidrogenación de iminas en 1997 encontrando los mejores resultados con catalizadores de Rh, Ir, Ru y ansa titanocenos, proponiendo para los sistemas basados en Rh o Ir el ciclo que se ve en la transparencia: adición oxidante de hidrógeno, inserción de imina en el enlace M-H dando el hidruro-amido complejoyelimi nación reductora N-H (más rápida que l a ß-eliminación como ya hemos comentado). La eficacia de los catalizadores está probablemente relacionada con la facilidad de formación del hidruro-amido complejo. Para el caso de los catalizadores de rutenio las cosas cambian.
Las reducciones asimétricas de dobles enlaces C=C y C=O mediante complejos de rutenio con agentes de transferencia de hidrógeno se han mostrado como una atractiva alternativa al uso de hidrógeno por el menor peligro y por la mayor simplicidad experimental. Sin embargo, su uso para la reducción enantioselectiva de dobles enlaces C=N estaba sin explorar hasta la publicación del grupo de Noyori (Premio Nobel 2001 compartido con Knowless y Sharpless) en 1996 utilizando como agente de transferencia de hidrógeno una mezcla de ácido fórmico y trietilamina y como catalizador un amidocomplejo de rutenio quiral, con el areno paracimeno (una diamina sulfonilada, el grupo paratoluensulfonilo parece clave tanto a la hora de estabilizar el amidocomplejo frente a la ß-eliminación como a la elevada reactividad ausente con otros grupos como bencilo o acetilo ). Se consiguen excesos enantioméricos elevados entre 90 y 97% con iminas cíclicas y un poco menores (85-89%) con iminas acíclicas.
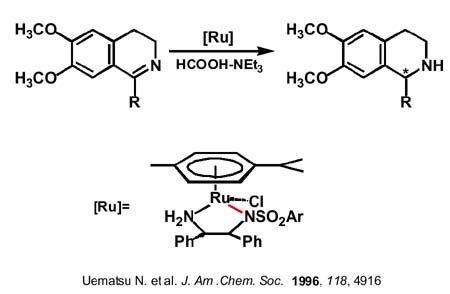
La clave del mecanismo por el que se lleva a cabo la hidrogenación de las iminas la podemos obtener al considerar que ese mismo amidocomplejo se muestra muy eficaz en transferencias de hidrógeno asimétricas entre alcoholes y cetonas. Por ejemplo, se obtienen rendimientos del 98% y excesos enantioméricos del 97% (próximos al 100%) hidrogenando acetofenona con alcohol isopropílico en medio básico aunque la especie que realmente cataliza es el amidocomplejo, que se ha podido caracterizar estructuralmente y que se forma in situ por adición del medio básico. La reacción del diamido con alcohol isopropílico produce el hidruro-amino el cual a su vez por reacción con gran exceso de acetona, regene-ra el diamido complejo. Podemos viajar en uno u otro sentido en condiciones suaves sólo cambiando la relación alcohol isopropílico-acetona.
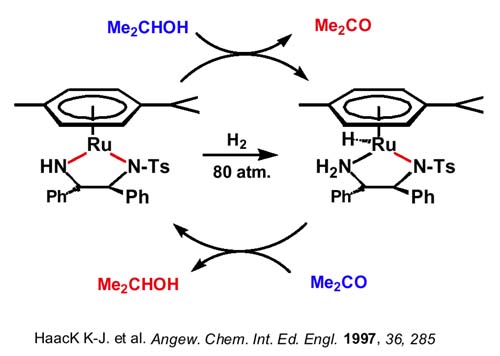
Nótese que la activación de la molécula de hidrógeno (que puede realizarse a presión) no se produce sobre el centro metálico en una adición oxidante (como en Rh o en Ir) sino sobre el enlace Ru-N pasando de amido a hidruro-amino lo que hace que no cambie el estado de oxidación del Ru(II) siempre (II).
Nota: Conferencia pronunciada por el Dr. Gabriel García Herbosa, durante el XXII Congreso Peruano de Química (octubre 2004). Continuará en el próximo número.
