




![Síntesis de nanopartículas de oro obtenidas por reducción de H[AuCl4]](/img/es/next.gif)





Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química del Perú
versión impresa ISSN 1810-634X
Rev. Soc. Quím. Perú v.78 n.2 Lima abr./jun. 2012
TRABAJOS ORIGINALES
Determinación de trazas de cadmio en presencia de morin-5´- ácido sulfónico como agente complejante, por voltamperometría de adsorción (AdSV) sobre electrodo de mercurio de gota colgante (HMDE)
Determination of traces of cadmium in the presence of morin-5´-sulphonic acid as complexing agent by adsorptive stripping voltammetry (AdSV) on hanging mercury drop electrode (HMDE)
Edgar Naglesa*, Verónica Arancibiaa, Roxana Ríosb, Manuel Otinianoc
a* Facultad de Química, Pontificia Universidad Católica de Chile, Santiago 7820436, Chile. e-mail: ernaagles@uc.cl.
b Facultad de Ciencias Químicas y Farmacéuticas, Universidad de Chile, Santiago, Chile.
c Facultad de Química e Ingeniería Química, Universidad Nacional Mayor de San Marcos, Perú.
RESUMEN
Un simple y sensible método electroanalítico es desarrollado para la determinación de cadmio por voltamperometría de adsorción (AdSV) en la presencia de morín-5´-ácido sulfónico (MSA). El complejo Cd (MSA) acumulado sobre la superficie del electrodo de mercurio de gota colgante (HMDE) y el pico de corriente es medido por voltametría de onda cuadrada (SWV). El complejo es reducido a -0,69 V. Las variables experimentales como pH, concentración de ligando (CMSA), tiempo de adsorción (tads) y potencial de adsorción (Eads), así como también las potenciales interferencias, fueron estudiadas. Bajo las óptimas condiciones (pH 3,9; CQSA 2,5 µmol L-1; tacc 80s; Eacc -0,30 V) el pico de la corriente es proporcional a la concentración de Cd(II) entre 0,1 to 12,0 µg L-1, con un límite de detección de 0,1 µg L-1. La desviación estándar relativa para una solución conteniendo 10,0 µg L-1 de Cd(II) fue de 1,08% para siete medidas iguales. El método fue validado por determinación de Cd(II) en agua de mar sintética (ASTM D665) dopada con ICP solución estándar de multielementos y en agua residual certificada (SPS-WW1). Finalmente, el método fue aplicado en la determinación de Cd(II) en muestras de agua potable, de mar y residual, después de ser atacadas con radiación UV para eliminar la materia orgánica.
Palabras clave: Cadmio; voltamperometría de adsorción; morín-5´-acido sulfónico.
ABSTRACT
A simple and sensitive electroanalytical method is developed for the determination of cadmium by adsorptive stripping voltammetry (AdSV) in the presence of morin-5'-sulphonic acid (MSA). The Cd (MSA)2 complex accumulates on the surface of a hanging mercury drop electrode (HMDE) and peak current is measured by square wave voltammetry (SWV). The complex is reduced at -0.69V. The experimental variables as pH, MSA concentration (CMSA), accumulation time (tacc), accumulation potential (Eacc), as well as potential interferences, are investigated. Under the optimized conditions (pH 3.9; CQSA 2.5 µmol L-1; tacc 80s; Eacc -0.30 V), peak current is proportional to the concentration of Cd(II) over the 0.1 to 12.0 µg L-1 range, with a detection limit of 0.1 µg L-1. The relative standard deviation for a solution containing 10.0 µg L-1 of Cd(II) solution was 1.08% for seven successive assays. The method was validated by determining Cd(II) in synthetic sea water (ASTM D665) spiked with ICP multielement standard solution and in certified reference water (SPS-WW1). Finally, the method was successfully applied to the determination of Cd (II) in tap water after UV digestion.
Key words: Cadmium, adsorptive stripping voltammetry, morin-5´-sulphonic acid.
INTRODUCCIÓN
La importancia de analizar iones de metales pesados se basa principalmente en su alta toxicidad y su rápida acumulación en algunos tejidos de los seres vivos, causando cáncer en la mayoría de los casos1.
Para realizar análisis de metales a nivel de trazas, la redisolución anódica (ASV) ha sido la técnica más utilizada en los últimos 50 años, debido a su bajo límite de detección (en algunos casos bajo µg L-1), precisión, exactitud y bajo costo de instrumentación e insumos. Los electrodos de gota de mercurio (HMDE) y de película de mercurio (MFE) han sido los más usados en esta técnica, gracias a su reproducibilidad y sensibilidad. Por esta técnica se han reportado varios trabajos para determinar cadmio con límites de detección entre 0,58 y 0,16 µg L-1 sobre (HMDE)2,3,4 y 0,005 µg L-1 sobre MFE5. Esta técnica se basa en una previa acumulación del analito, debido a una reacción de electrólisis y posterior oxidación, realizando el barrido con alguna técnica moderna como voltamperometría diferencial de pulso (DPV) o voltamperometría de onda cuadrada (SWV). Estas dos etapas aportan gran sensibilidad a la técnica.
Otra alternativa más reciente es el empleo de la voltamperometría de adsorción (AdSV). En esta técnica la etapa de acumulación ocurre por un proceso de adsorción del analito en el electrodo de trabajo. Debido a que los iones metálicos solvatados no se adsorben con facilidad en los electrodos de trabajo, es necesario que éste se encuentre formando un complejo estable con un ligando conveniente. El ligando, al tener grupos -OH; -SH; -COO, etc., hace que el complejo pueda acumularse en la superficie del electrodo de acuerdo a diversos mecanismos.6 Existen buenas referencias con los fundamentos y aplicaciones de la AdSV; cabe mencionar los trabajos de Voulgaropoulos6, Zuhri7, Kalvoda8,9, Economou10, Honeychurch11, Abollino12, Van den Berg13. Algunos ligandos usados en AdSV para determinar Cd(II) son: 8 hidroxiquinolona3,4, 2,7(PADN), CNAQS15, 5-hidroxiquinolona16, 2,5-DMT2, quercetin17,18. Este trabajo apunta hacia el uso del flavonoide morín con derivado sulfúrico como agente complejante de Cd(II), el cual ha sido usado para determinar Pb(II) sobre HMDE19 y muestra un pico de corriente de la reducción de Pb(II) a un potencial diferente del pico de corriente de la reducción de Cd(II).
PARTE EXPERIMENTAL
Reactivos
El agua usada para lavar y preparar disoluciones fue agua desionizada 18 MΩ cm de un sistema milliQ o agua destilada calidad HPLC (Merck). Las disoluciones patrones con 1,0 mg L-1 Cd(II) fueron preparadas a partir de patrones Titrisol (Merck) de 1000 mg L-1. El ácido morín-5´-ácido sulfónico fue sintetizado como es reportado por Kopack20 y fue preparado en metanol. Las disoluciones de tampones Britton-Robinson (BR) fueron preparadas por mezcla de iguales volúmenes de ácido fosfórico, ácido acético y ácido bórico 0,4 mol L, ajustando el pH requerido con disolución de NaOH 2,0 mol L-1 y diluyendo 10 veces con agua. Se utilizó agua de mar sintética ASTM D665 (Aldrich) y Estándar Certificado de agua residual SPS-WW1.
Instrumentos y equipos
Para el desarrollo de las medidas voltamperométricas se utilizó un polarógrafo Radiometer (Lyon Frace) POL-150, con el programa Trace-Master 5 PC para la adquisición de datos. La celda consta de un sistema de tres electrodos; un electrodo de trabajo de gota colgante de mercurio (HMDE), un electrodo de referencia de Ag/AgCl/ KCl 3 mol L-1, un alambre de platino como electrodo auxiliar, cánula para el paso de argón y barra para agitación. Para la eliminación de la materia orgánica de las muestras reales se utilizó el digestor Metrohn 705 UV con lámpara de mercurio. En la preparación de los tampones se utilizó un pH-metro Orion 430 con electrodo de membrana de vidrio.
Procedimiento
En la celda electroquímica se agregó 10,0 mL de agua desionizada 18 MΩ cm y 500 µL de tampón; se desgasa la solución con gas argón por 10 minutos; se trazó el blanco. Posteriormente se agregó el ligando entre 0,1 y 10 µmol L-1 y se trazó el respectivo voltamperograma del ligando; después se adicionaron alícuotas de Cd(II) entre 4,9 y 9,8 µgL -1 cuando se buscaban los parámetros óptimos. En las curvas de calibrado se adicionó la misma cantidad de agua desionizada y tampón.
RESULTADOS Y DISCUSIÓN
Estudio en función del pH de la disolución
Uno de los parámetros más importantes es el pH de la disolución. La constante de equilibrio de complejación cambia y además, como el ligando tiene diferentes grupos ácidos, la carga neta del complejo también cambia. En este estudio es más importante la carga del complejo que el valor de la constante de equilibrio. Una constante pequeña se minimiza agregando el ligando en exceso.
En este estudio se realizó medidas con 4,9 µg L-1 Cd(II), MSA 1,8 µmol L-1 y se varió el pH de la disolución entre 2,0 y 7,0 utilizando 100 µL de tampones BR de concentración 0,4 mol L-1. Se aplicó tads: 60 s; Eads -0,20 V; amplitud del escalón de 10 mV; amplitud del pulso 50 mV y frecuencia de la onda cuadrada de 25 Hz. Los resultados se presentan en la figura 1.
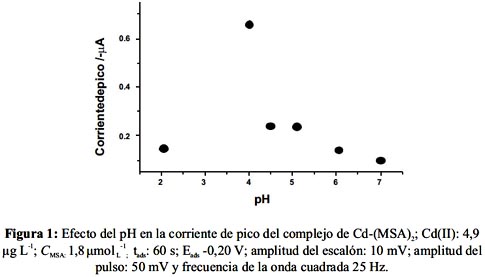
Como se muestra, la mayor corriente de pico se obtuvo a pH 3,9 y luego a pH mayores decrece rápidamente. Éste fue el valor escogido para todas las mediciones posteriores. Por otra parte, la señal del complejo Cd(MSA)2 se desplaza a potenciales más negativos al aumentar el pH.
Estudio en función de la concentración de ligando (CMSA)
El ácido morín-5-sulfónico (MSA) cuyo nombre IUPAC es (3,5,7,2´,4´-pentahidroxi-5´sulfoflavona) forma un complejo con Cd(II) de estequiometria metal:ligando 1:221. ApH entre 4-6 el ligando está en forma dianiónica, por lo que se obtendría un complejo con carga negativa, [Cd(H3L)22-]. Para este estudio se utilizó una disolución de Cd(II) 4,9 µg L-1 y se varió la concentración de ligando entre 0,0 y 3,5 µmol L-1 (en disolución). Las otras condiciones fueron: pH: 3,0; tads: 60 s; Eads -0.20 V; amplitud del escalón: 10 mV; amplitud del pulso: 50 mV y frecuencia de 25 Hz. Los resultados obtenidos se presentan en las figuras 2.
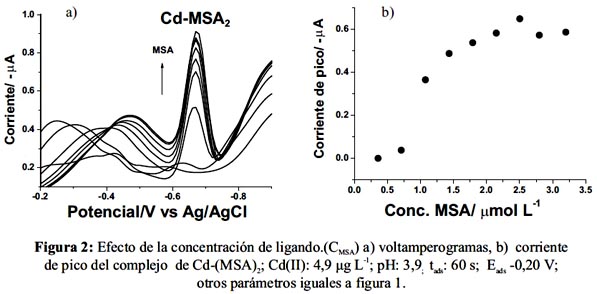
Como muestra la figura 2 (a), la corriente de pico aumenta bruscamente con la concentración de ligando desde 0,7 a ≈ 2,5 μmol L -1 y luego se mantiene constante. Para las medidas posteriores se eligió un valor de 2,5 μmol L-1 de MSA óptima.
Estudio en función del potencial de adsorción (Eads)
Este estudio se realizó utilizando una disolución de Cd(II) 4,9 µg L-1 y se varió el potencial de adsorción entre 0,00 y -0,55 V, mientras las otras condiciones permanecieron constantes: pH 3,9; CMSA 2,5 μmol L-1 ; tads 60 s; amplitud del escalón: 10 mV; amplitud del pulso: 50 mV y frecuencia de 25 Hz. Los resultados obtenidos se presentan en la figura 3.
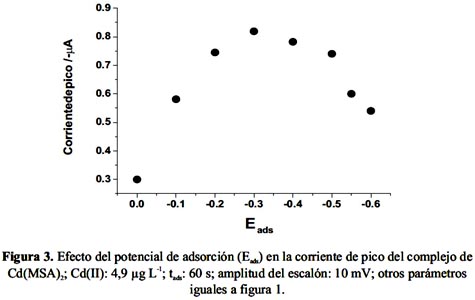
Como muestra la figura 3, la corriente de pico aumenta hasta un potencial de adsorción de –0,30 V para luego decrecer bruscamente. El valor de -0,30 V fue elegido para las posteriores mediciones.
Estudio en función del tiempo de acumulación (tads)
Este estudio se realizó utilizando una disolución de Cd(II) 4,9 µg L-1 y se varió el tiempo de adsorción entre 0-150 s mientras las otras condiciones permanecieron constantes: pH 3,9; CMSA 2,5 μmol L-1 Eads-0,30 V; amplitud del paso: 10 mV; amplitud del pulso: 50 mVy frecuencia de 25 Hz. Los resultados obtenidos se presentan en la figura 4.
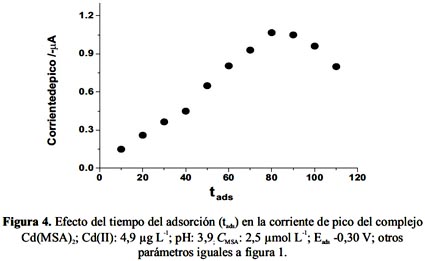
Como se muestra en la figura 4, la corriente de pico aumenta con el tads hasta 80 s y luego decrece ligeramente. El tads escogido fue 80s.
Parámetros de la técnica en la etapa de barrido
Al igual que en los estudios anteriores se eligió un tamaño de gota de mercurio de 0,75 mm2, una frecuencia de 25 Hz, un potencial del escalón de 10 mV y una amplitud del pulso de 50 mV. A otros valores diferentes se pierde la forma del pico de reducción.
Construcción de la curva de calibrado
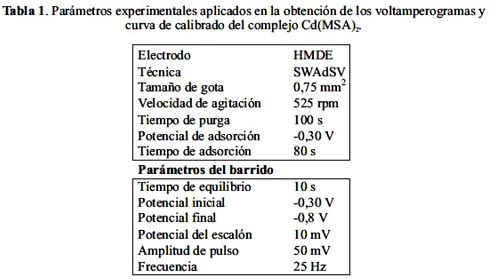
Los voltamperogramas de adsorción, en función de la concentración de Cd(II), fueron obtenidos en las siguientes condiciones experimentales: pH 3,9 (100 μL de tampón BR 0,4 mol L-1); CMSA : 2,5 μmol L-1 ; tads: 80s y Eads: -0,30 V. En la tabla 1 se resumen los parámetros experimentales aplicados y en la figura 5 los resultados obtenidos.
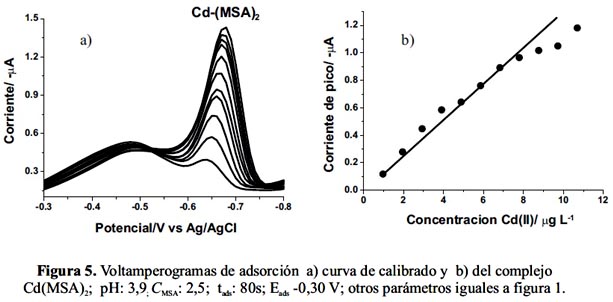
Si CMSA es de 2,5 μmol L-1 la curva de calibrado del complejo Cd-(MSA) tiene un rango de linealidad de 0,0-11,0 μg L-1. La precisión expresada como la desviación estándar relativa (R.S.D) fue de 1,08 % para siete medidas consecutivas de la misma muestra conteniendo 4,9 μg L-1 de Cd(II). El límite de detección (LD) según el método Milller and Miller22 fue de 0,10 ± 0,8 µg L-1.
Validación de la metodología con agua de mar dopada y agua certificada
Agua de mar sintética (ASTM D665)
Para realizar la validación se utilizó agua de mar comercial (ASTM D665) que contiene la misma matriz de una muestra de mar real, sólo se dopó con Cd(II). Auna alícuota de 1,0 mLde agua de mar se agregó agua desionizada y 100 µL de tampón BR. La concentración de Cd(II) en disolución fue de 6,9 μg L-1 (dopada). El valor obtenido al utilizar el método de la adición de estándar fue de 6,5 ± 0,1 µg L-1 (ER: -5,79 %)
Estándar certificado SPS-WW1
La validación también se realizó con agua residual certificada (SPS-WW1) la cual contenía: Al 2000; As 100; Cd 20.0; Co 60; Cr 200; Cu 400; Fe 1000; Mn 400; Ni 1000; P 1000; Pb 100; V 100 and Zn 600 µg L-1. Se adicionó 1000 µL de muestra, lo cual significa que en disolución hay 2,0 µg L-1 de Cd(II). Las condiciones experimentales fueron: pH 3,9 (BR); CMSA: 3,0 μmol L-1; tads: 80 s; Eads: -0,30 V. Este tipo de matriz permitió evaluar también las posibles interferencias de otros iones metálicos en la determinación de Cd(II).
Al utilizar el método de la adición de estándar se obtuvo un valor de 1,8 µg L-1 de Cd(II) (ER: 10 %). Estas mediciones se realizaron por triplicado. Esto indica que otros iones metálicos como Cu(II), Ni(II), Pb(II), y Zn(II) los cuales se encuentran en concentración de 8, 20,0, 0,4 y 12,0 μg L-1 en disolución respectivamente, no interfieren. En muestras reales de agua potable no se detectó Cd(II). Estas muestras se analizaron en un laboratorio de servicios (por ICP-AES) en el cual se informó un valor inferior a 0,01 μg L-1.
CONCLUSIONES
El método propuesto es conveniente para determinar Cd(II) en matrices de aguas naturales y aguas residuales. La presencia de otros iones metálicos no interfiere en el análisis pero consumen MSA por la capacidad que tienen de formar complejos con otros iones metálicos.
AGRADECIMIENTOS
Los autores agradecen a FONDECYT por el financiamiento del Proyecto Postdoctorado 3120030
BIBLIOGRAFÍA
1. Manahan S.E. Introducción a la Química Ambiental. Editorial Reverté UNAM, México, Pág. 148-149,529. 2007.
2. Van den Berg C.M.G. J. Electroanal. Chem. Acta 1986; 215:111.
3. Surer E.; Vilmaz S.; Turkoglu M. Environ. Monit. Assess. 2007; 125: 91.
4. Abollino O.; Aceto M.; Sacchero G.; Sarzanini C.; Mentasi E. Anal. Chim. Acta, 1995; 305: 200-206.
5. Fischer E.; Van den Berg C.M.G. Anal. Chim. Acta 1999; 385: 273-280.
6. Panelli M.G.; Voulgaropoulos A., Electroanalysis 5: 355, 1993 Zuhri A. Z. A.; Voelter W. Fresenius J. Anal. Chem. 1998; 360: 1.
7. Kalvoda R. Fresenius J. Anal. Chem. 1994; 349: 565.
8. Kalvoda R. Anal. Chim. Acta 1984; 162: 197.
9. Economou A.; Fielden P.R.; Anal. Chem. 1997; 16: 286.
10. Honeychurch M.; Rechnitz G.A. Electroanálisis. 1998; 10: 285.
11. Abollino O.; Aceto M.; Zarzanini C.; Mentasti E. Electroanalysis 1999;11: 870.
12. Van den Berg C.M.G. Anal. Chim. Acta. 1991; 250: 265.
13. Zhang Z. Q.; Chen S. Z; Lin H. M.; Zhang H. Anal. Chim. Acta 1993; 272: 227.
14. Ornella A, Mauricio A, Giovanni S, Corrado S, Edoardo M, Anal Chim. Acta. 1995; 305: 200.
15. Si Q.; Xu S.; Cai J.; Lu R.; Zhu Y. Fenxi Huaxue. 1992; 272.
16. Li C.; James B.D.; Rumble J.; Magee R.J. Mikrochim. Acta III 1998; 175.
17. Farghly O.; Wadood H.; Mohamed H. J. Pharmaceutical Sci. 2003; 17: 43.
18. Arancibia V.; Nagles E.; Cornejo Sh. Talanta. 2009; 80: 184.
19. Kopacz M. Polish J. Chem. 1981; 55: 227-229.
20. Kuzniar A.; Kopacz M.; Nowak D. J. Coordination Chem. 2008; 61: 1005-1018.
21. Miller J.C.; Miller J.N. Estadística y Quimiometría para Química Analítica, Addison-Wesley, Cuarta edición; 124-125; 2002.
Recibido el 04-04-2012
Aprobado el 18-04-2012
