










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química del Perú
versión impresa ISSN 1810-634X
Rev. Soc. Quím. Perú vol.80 no.2 Lima abr. 2014
TRABAJO ORIGINAL
Estudio de la reactividad en moléculas fenólicas mediante la función de Fukui
Study of reactivity in phenol molecules through the Fukui function
Carlos A. Sernaqué1; Luis G. Calvo1; Rodolfo Pumachagua1.*
1 Universidad Nacional Federico Villarreal, Facultad de Ciencias Naturales y Matemáticas, Laboratorio de Química Teórica, Jr. Chepen s/n (El Agustino) Perú. rpumachagua@yahoo.es
RESUMEN
En este trabajo analizamos la estructura más estable y la evolución de la reactividad local utilizando los índices de Fukui en sistemas fenólicos sustituidos en posición-para, conforme se modifica la función base en diferentes métodos. Los resultados explican las sustituciones: electrofílica y nucleofílica aromática y la acilación en los sistemas fenólicos. Los cálculos se llevaron a cabo a un nivel: HF/3-21G, B3LYP/3-21G, B3LYP/6-31G(d) y MP2/6-31+G(d); se estableció el nivel de cálculo adecuado para cada tipo de reacción estudiada.
Palabras clave: Teoría del funcional de densidad, función de Fukui, fenol, carácter nucleofílico.
ABSTRACT
In this paper we analyze the most stable structure and the evolution of the reactivity of local using the indices of Fukui in systems phenolic substituted in position-para as amending function based on different methods. The results explain the substitutions: Electrophilic and aromatic nucleophilic and acylation in phenolic systems. The calculations were carried out at a level: HF/3-21G, B3LYP/3-21G, B3LYP/6-31G(d) and MP2/6-31+G(d), established the level of calculation for each type of reaction studied.
Key words: Functional of density theory, Fukui function, phenol, character nucleophilic.
INTRODUCCIÓN
Los fenoles contienen un grupo hidroxilo unido directamente a un grupo aromatico1, son sustratos reactivos a la sustitución electrofílica aromática porque el grupo hidroxilo dona electrones al anillo que estabiliza al carbocatión intermediario formado; el efecto de resonancia dirige la reacción en posición orto y para.
La reacción de nitración del fenol es 1000 veces más rápido que del benceno2. La sustitución nucleofílica del fenol ocurre en presencia de un grupo saliente atractor de electrones2. La acilación de los fenoles en el oxígeno del hidroxilo ocurre bajo condiciones especiales de Friedel-Craft, sujetas a un control cinético y termodinámico1.
Analizamos las reacciones de sustituciones: electrofílica y nucleofílica cuando existe un sustituyente en posición para respecto al grupo hidroxilo del fenol por ser uno de los sitios de mayor densidad electrónica3; de esta manera, podemos explicar los sitios reactivos locales con la función de Fukui para obtener un solo producto de reacción en los dos tipos de sustitución aromática, deseado hoy en día por los laboratorios de investigación4.
Los cálculos de reactividad local mediante la función de Fukui se obtuvo a partir de las cargas de Mulliken, obtenidas de los diferentes cálculos de optimización. De esta manera encontramos el método que prediga la energía más baja para nuestro sistema y determine los sitios de mayor reactividad1, 2.
FUNDAMENTOS TEÓRICOS
Método Moller-Plesset
Basada en la teoría de perturbación, el Hamiltoniano se expresa como5, 6:

donde H(0) es el hamiltoniano de orden cero del cual se conocen sus funciones propias y valores propios. H es el operador de perturbación.
Mediante el parámetro λ, que va de 0 (sistema sin perturbar) a 1 (perturbación aplicada completamente)5 se construye una serie de Taylor para encontrar los niveles energéticos En y sus respectivas funciones propias, Ψn que se pueden expresar de la forma:
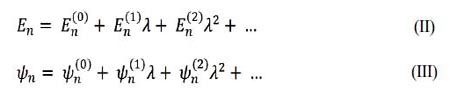
donde Ψn(K) y En(K) son las correcciones de orden K de la función de onda y de la energía n n respectivamente. Por lo tanto, para encontrar las correcciones se utiliza la ecuación de Schrödinger independiente del tiempo5, 6:
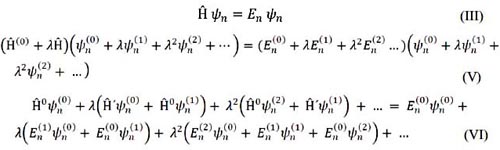
Igualando términos:
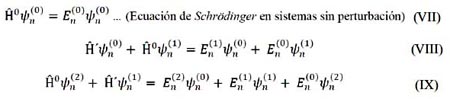
Las correcciones de energía de primer y segundo orden se expresan6:
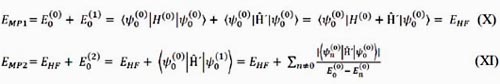
De esta manera, el método MP2 incluye la primera correlación electrónica de menor coste computacional, y por ello es uno de los más utilizados de los métodos perturbacionales.
Cabe indicar, que los métodos perturbacionales son consistentes con el tamaño, lo que combinado con su menor coste computacional respecto a los métodos de Interacción Configuracional, los hace los métodos más utilizados de correlación electrónica6.
Función de Fukui
Es una propiedad local que se describe como la primera derivada de la densidad electrónica ρ(r) de un sistema con respecto al número de electrones N a un potencial externo constante v(r). Esto se expresa como7, 8:
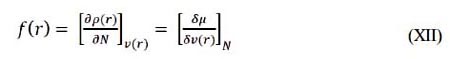
Las funciones de Fukui nucleofílica, electrofílica y radical se expresan como7, 8:

donde ρk (N), ρk (N-1) y ρk (N+1) son las poblaciones electrónicas del sitio k en los sistemas neutros, catiónicos y aniónicos, respectivamente7.
ASPECTOS COMPUTACIONALES
Los cálculos computacionales se realizaron utilizando el programa Gaussian 03. Las optimizaciones de geometría se llevaron a cabo al nivel teórico: HF/3-21G, B3LYP/3-21G, B3LYP/6-31G(d) y MP2/6-31+G(d).
El estudio de las funciones de Fukui se realizó a partir del cálculo puntual9 para los sistemas catiónicos y aniónicos variando la multiplicidad y la carga, manteniendo la geometría del sistema neutro7.
RESULTADOS Y DISCUSIÓN
El fenol sustituido en posición para, sufre un ataque electrofílico en las posiciones 2 o 6 (figura 1). Las posiciones 2 y 6 son posiciones orto con respecto al activador hidroxilo del núcleo fenólico, que tiene preferencia sobre otros sustituyentes a excepción del grupo amino.
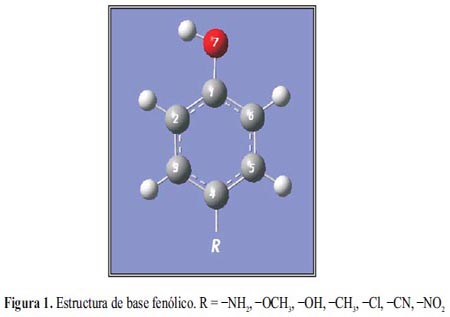

Para un ataque nucleofílico, la reacción ocurre en las posiciones 1 o 4 (figura 1) que presentan menor densidad electrónica con respecto a los demás carbonos1, 2.
La tabla 2 resume los sitios de reacción para una sustitución electrofílica y nucleofílica de cada molécula en estudio.
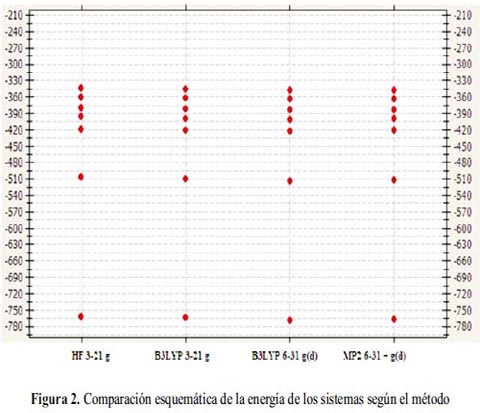
Los valores en la tabla 3 muestran que el método de la teoría funcional de densidad (DFT): B3LYP/6-31G(d) proporciona la energía más baja para cada una de las moléculas en estudio (tabla 1), y que la función base: 6-31G(d) describe adecuadamente a cada molécula fenólica, en lugar de la función base: 3-21G.
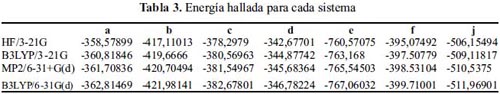
El método de la teoría de perturbación Moller-Plesset: MP2/6-31+G(d) se encuentra próximo en valores de energía.
La figura 2 muestra la tendencia de la energía para cada una de las moléculas en función del método teórico.
Tablas de reactividad
Nivel de teoría HF/3-21G
Observando la tabla 4, el ataque por una especie electrofílica sucede en el carbono 1 o 4, porque presentan mayor densidad electrónica. Este resultado no es viable teóricamente
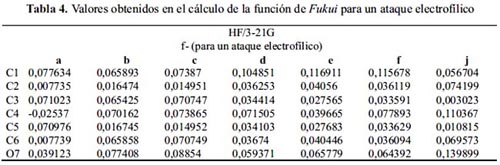
Por otro lado, los sistemas: b, c y j –este último posee el valor más alto en reactividad a un ataque electrofílico– tienen en el átomo 7 el sitio más reactivo a un ataque electrofílico; los electrones libres del oxígeno determinan su capacidad donadora, como sucede en la reacción de acilación de fenoles. Sin embargo, el método es inadecuado para calcular adecuadamente la función de Fukui f-.
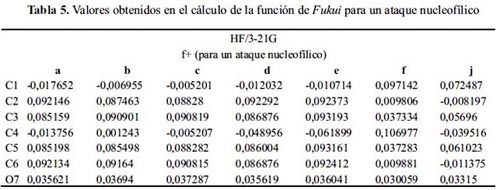
Además, la tabla 5 muestra que el ataque por una especie nucleofílica para todas las moléculas sucede en cualquier carbono diferente a 1 y 4, excepto en las moléculas: f y j.
Nivel de teoría B3LYP/3-21G
De la tabla 6 para el ataque electrofílico, observamos uniformidad en los valores de la función de Fukui para todos los sistemas, mayores que con el método de Hartree-Fock.
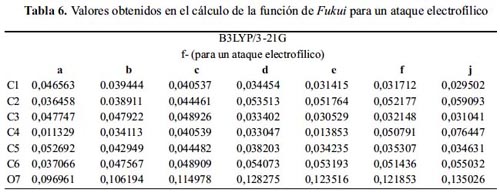
El sitio más reactivo en todos los sistemas es el átomo 7 –correspondiente al átomo de oxígeno– donde se concentra la mayor densidad electrónica del sistema, y dispuesto a reaccionar con un electrófilo. Este método explica de manera homogénea la acilación de los fenoles.
Si tomamos al segundo átomo más nucléofilo de las moléculas: d, e y f, reaccionan con una especie electrofílica en los C2 o C6. Asimismo, la molécula b reacciona con una especie electrofílica en el C3 y la molécula a en el C5, es decir, en posición orto con respecto al grupo amino por su mayor carácter activador sobre el grupo hidroxilo. La sustitución electrofílica en los sistemas: a, b, d, e y f es coherente con la teoría.
Observando los valores de la tabla 7, la reacción de las moléculas: e y f con una especie nucleofílica sucederá en el carbono 4, teóricamente válida. Las demás moléculas no logran ser explicadas mediante este método para una sustitución nucleofílica.
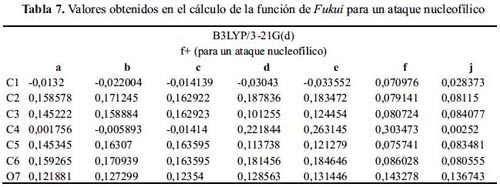
Nivel de teoría B3LYP/6-31G(d)
En la tabla 8 observamos que todos los sistemas, a excepción de la molécula d, presentan el sitio más nucleófilo en el átomo 7, teniendo la molécula j el valor más alto. La molécula d presenta el sitio más reactivo a un ataque electrofílico en el C2, que concuerda con los datos teóricos. Si tomamos el segundo valor más alto del índice de Fukui nucleofílico para las moléculas: d y e, la sustitución electrofílica se daría en el C2, acorde con la teoría.
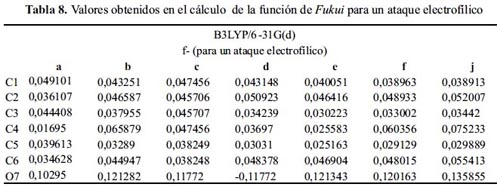
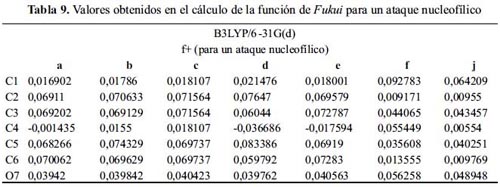
La tabla 9 indica que la reacción de las moléculas: f y j con una especie nucleofílica sucede en el C1. A pesar de ser uno de los sitios con menor densidad electrónica, no es posible que ocurra la reacción en ese lugar. Los grupos: ciano y nitro, por efectos inductivos, la reacción se daría en el C1.
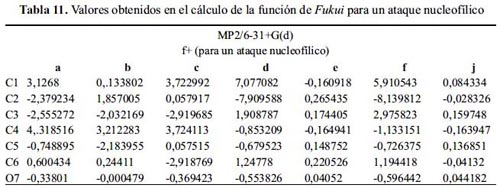
Nivel de teoría MP2/6-31+G(d)
La tabla 10 muestra mayor variedad en la reactividad para el ataque electrofílico que en los métodos anteriores. Las moléculas: d, e y j presentan como sitio nucleófilo al O7, justificando la reacción de acilación de los fenoles, mientras que las moléculas: b y c justifican satisfactoriamente la sustitución electrofílica en el C2.
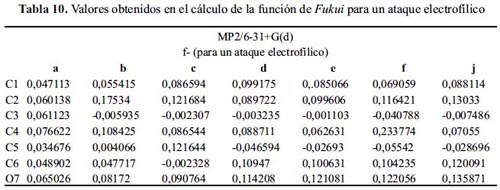
Considerando el segundo mayor índice de Fukui nucleofílico (tabla 11), la reacción con un electrófilo para las moléculas: d, e y j sucedería en el carbono 2 o 6, como predice la teoría. El ataque de un nucleófilo a las moléculas: a, b y c sucede en C4, y en las moléculas: d y f suceden en C1.
La tabla 12 resume los mejores resultados para explicar la reactividad nucleofílica/electrofílica de las moléculas fenólicas sustituidas en posición para. El método MP2/6-31+G(d) destaca entre los métodos utilizados.

CONCLUSIONES
Los métodos: B3LYP/3-21G(d) y B3LYP/6-31G(d) explican adecuadamente la reacción de acilación en los sistemas fenólicos sustituidos en posición para.
Los métodos: B3LYP/3-21G(d) y MP2/6-31+G(d) describen correctamente la sustitución electrofílica.
La sustitución nucleofílica determinada por el índice de Fukui para cada método fue variado, siendo los mejores: B3LYP/3-21G(d) y MP2/6-31+G(d).
El método B3LYP/3-21G(d) genera los valores de energía más bajos para los sistemas estudiados, y se complementa con el método MP2/6-31+G(d) para describir el índice de Fukui para la sustitución electrofílica y nucleofílica.
AGRADECIMIENTO
Los autores agradecen al Dr. William Tiznado Vásquez de la Universidad Andrés Bello, Santiago de Chile, por brindarnos soporte computacional.
REFERENCIAS
1. Francis A. Carey, Química Orgánica. Sexta edición. Mc Graw Hill, 2006, 1005-1017.
2. John McMurry, Química Orgánica. Séptima edición. Cengage Learning, 2008, 560-574.
3. Patricia Pérez, Luis R. Domingo, Mario Duque-Noreña & Eduardo Chamorro. A condensed-to-atom nucleophilicity index- An application to the director effects on the electrophilic aromatic substitutions. Journal of Molecular Structure:THEOCHEM, 2009; 895: 86-91.
4. Luis R. Domingo & Patricia Pérez. The nucleophilicity N index in organic chemistry. Org. & Biomol. Chem., 2011; 9: 7168-7175.
5. Ira N. Levine, Química Cuántica, Prentice Hall, 5ta edición, 2001, 242-248, 544-549
6. Joan Bertran, Vicenc Branchadell, Miquel Moreno & Mariona Sodupe, Química Cuántica, Síntesis S.A. 2da edición, 2002, 75-82, 193-198.
7. A. Ghomri & S. M. Mekelleche. Reactivity and regioselectivity of five-membered heterocycles in electrophilic aromatic substitution: A theoretical investigation. Journal of Molecular Structure: THEOCHEM, 2010; 941: 36-40.
8. Virginia Popa. La cuantificación de los sitios activos en las bases de DNA y RNA utilizando las funciones de Fukui condensadas. Revista Mexicana de Física, 2007; 53 (4): 241-253.
9. C. V. Uribe, E. A. Tobón, F. Figueroa & D. P. Henao. Fototoxicidad de medicamentos sulfas y su reactividad frente al oxígeno molecular singulete, Rev. Soc. Quím. Perú, 2008; 74 (4): 311-322.

