










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de la Sociedad Química del Perú
Print version ISSN 1810-634X
Rev. Soc. Quím. Perú vol.81 no.1 Lima Jan./Mar. 2015
TRABAJOS ORIGINALES
Evaluación teórica de nuevos derivados nitratoxicarbono de tetraedrano
Theoretical evaluation of new derivatives of the tetrahedrane nitratoxycarbon
Luis G. Calvo1, Rodolfo Pumachagua1*
1
Universidad Nacional Federico Villarreal, Facultad de Ciencias Naturales y Matemáticas, Laboratorio de Química Teórica, Jr. Chepen s/n (El Agustino) Perú. rpumachagua@gmail.com
RESUMEN
Se ha realizado el estudio teórico de derivados nitratoxicarbono de tetraedrano empleando los métodos: Hartree-Fock y la Teoría del funcional de la densidad. Determinamos la estructura, las propiedades electrónicas y el perfil energético; así también el análisis poblacional de Mulliken para mostrar que el incremento de grupos nitratoxicarbono disminuye las cargas de los oxígenos del grupo sustituyente.
Palabras clave: Nitratoxicarbono, tetraedrano, hidrocarburo platónico, B3LYP.
ABSTRACT
We calculated the properties of nitratoxycarbon tetrahedrane derivatives using the methods: Hartree-Fock and density functional theory. We assessed structure, the electronic properties and energy profile to show Mulliken population analysis that the increase of nitratoxycarbon groups reduce the burdens of the oxygens of the group substituent.
Key words: Nitratoxycarbon, tetrahedrane, platonic hydrocarbon, B3LYP.
INTRODUCCIÓN
El tetraedrano1,2 (figura 1) es un hidrocarburo platónico con cuatro átomos de carbono formando los vértices de un sistema tetraédrico. El esqueleto de carbono encierra una cavidad en forma de jaula, el ángulo de 60 grados permite almacenar una gran cantidad de energía en estos enlaces. Es interesante por al menos cuatro razones: es un hidrocarburo platónico, es una prueba del límite estructural en la química orgánica, constituye un desafío para la síntesis y es un ejemplo de química exótica3,4.
Los materiales con alta capacidad energética son los compuestos en forma de jaula con alta tensión angular y torsional; al sustituir sus hidrógenos por grupos nitrados obtienen propiedades explosivas5 muy potentes, como el tetranitrocubano6-9 o el trinitrotolueno. Por tanto, es importante conocer las propiedades electrónicas y estructurales previas a la síntesis10.
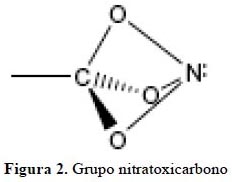
Diversos derivados nitro sobre el tetraedrano han sido estudiados11. El grupo nitratoxicarbono12 mostrado en la figura 2 ha sido planteado recientemente13. Por otro lado, el grupo nitratoxicarbono y los grupos nitro presentan características similares: el intercambio electrónico en los átomos de nitrógeno y oxígeno puede ser un factor estabilizante en los sistemas donde ellos participan; así también, cuando se incrementa el número de los sustituyentes (figura 3).
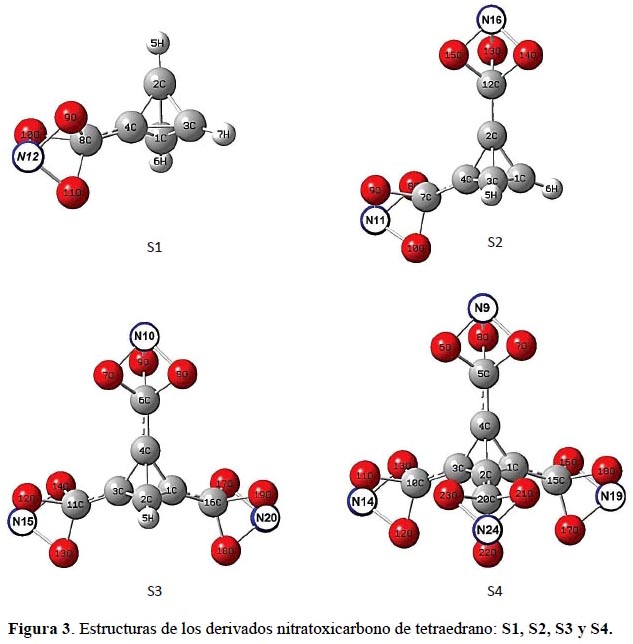
El interés en estos sistemas es la presencia del grupo nitratoxicarbono dentro de la molécula tetraedrano, las dimensiones del enlace, así como el perfil energético de la molécula y las frecuencias vibratorias permitirán plantear su estabilidad. Reportamos los resultados empleando los métodos Hartree-Fock (HF) y la de la Teoría del funcional de la densidad (DFT) empleando la función base 6-31G.
ASPECTO COMPUTACIONAL
Todos los cálculos se llevaron a cabo con el programa GAUSSIAN 09 usando la función base 6-31G con los métodos: Hartree-Fock y DFT utilizando el funcional híbrido B3LYP. Las frecuencias vibratorias se calcularon para cada molécula con ambos métodos para obtener estructuras estables.
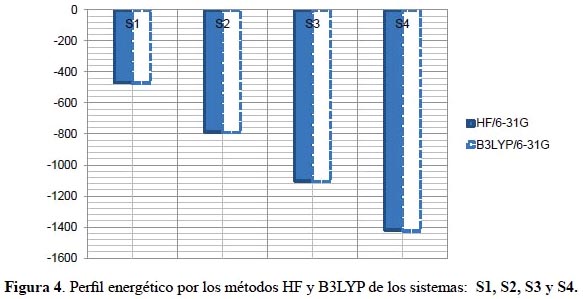
Perfil energético
Análisis poblacional de Mulliken
Propiedades estructurales
Propiedades electrónicas
RESULTADOS Y DISCUSIÓN
Perfil energético
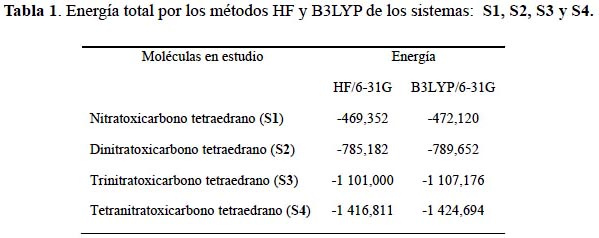
La geometría de equilibrio de las moléculas viene definido por una serie de coordenadas de los núcleos que permiten alcanzar un mínimo energético. Los sistemas derivados nitratoxicarbono del tetraedrano S1, S2, S3 y S4 en la tabla 1 muestran un descenso considerable de energía; la energía del S4 es la más baja de todos; observamos que mientras mayor es la cantidad de sustituyentes, la energía disminuye en ambos métodos de cálculo. Los valores más bajos son determinados por la teoría del funcional de la densidad.
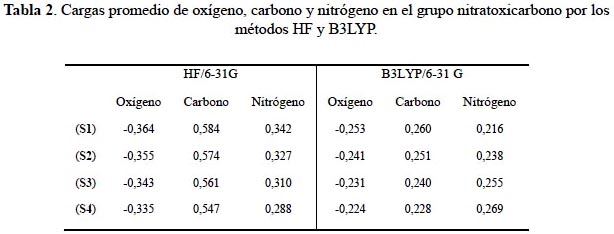
Análisis poblacional de Mulliken
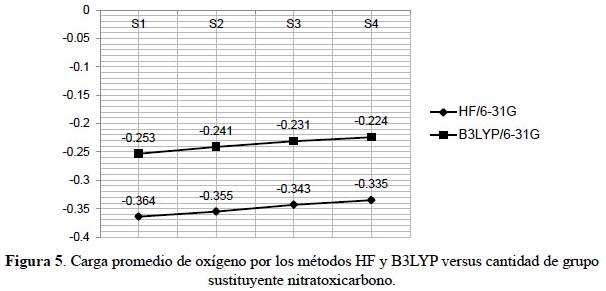
El análisis poblacional de Mulliken es un esquema arbitrario para asignar cargas de los átomos que forman una molécula. De la tabla 2 observamos una ligera disminución de carga negativa en los átomos de oxígeno conforme aumenta el número de sustituyentes nitratoxicarbono en el tetraedrano. El método Hartree-Fock presenta los valores de carga más bajos para el átomo de oxígeno. La pérdida de carga negativa en el oxígeno reduce las repulsiones electrostáticas entre ellos, dentro del sistema tetraédrico estableciendo las conformaciones más estables de los distintos sistemas.
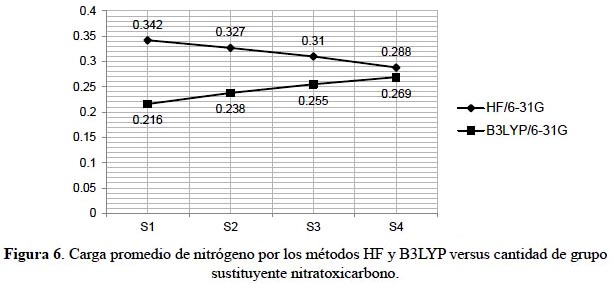
El átomo de nitrógeno muestra un comportamiento distinto. El método Hartree-Fock presenta una disminución de carga, ocasionando un ligero desequilibrio en las atracciones electrostáticas, porque tanto el oxígeno como el nitrógeno están perdiendo carga. Sin embargo, en DFT hay un incremento en la carga del nitrógeno que no afectaría en gran medida a las atracciones electrostáticas nitrógeno-oxígeno por una redistribución de la carga. El átomo de carbono une el sistema tetraédrico y el sustituyente muestra un comportamiento igual que los átomos de oxígeno en los dos métodos de cálculo.
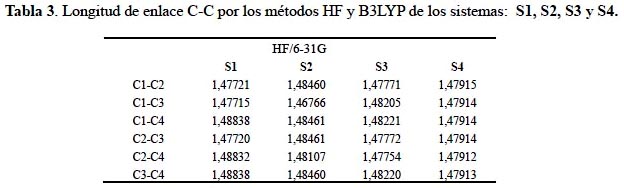
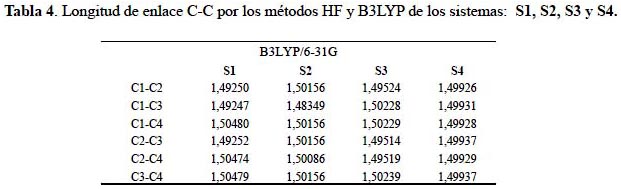
Propiedades estructurales
La geometría molecular es un indicador de las interacciones intra e inter-moleculares de cualquiera estructura química. En las tablas 3 y 4 observamos que al aumentar los grupos sustituyentes nitratoxicarbono hay un equilibrio significativo en las longitudes de enlaces del tetraedrano; dicho comportamiento se puede observar con cada sustitución.
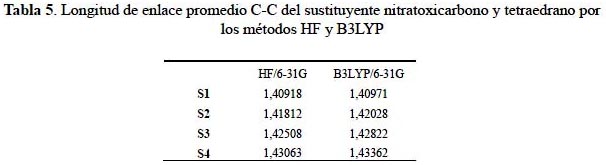
En la tabla 5 se observa que el enlace carbono-carbono entre sustituyente y el sistema tetraedrano se va alargando conforme aumenta el número de sustituciones. Este comportamiento se confirma por ambos métodos de cálculo, desde la longitud de 1,40971Å a 1,43362 Å con el funcional híbrido B3LYP y de 1,40918Å a 1,43063 Å con el método Hartree-Fock.
Las longitudes de enlace carbono-carbono obtenidas en los derivados nitratoxicarbono del tetraedrano mantienen valores muy cercanos, a diferencia de los valores obtenidos sobre la geometría molecular del tetranitrotetraedrano reportado en la literatura11. La introducción de los grupos nitro debilita los enlaces en el tetraedrano, incrementando la longitud en algunos enlaces carbono-carbono, llegando a medir 1,773Å y 1,770 Å, ocasionando la ruptura de los enlaces.
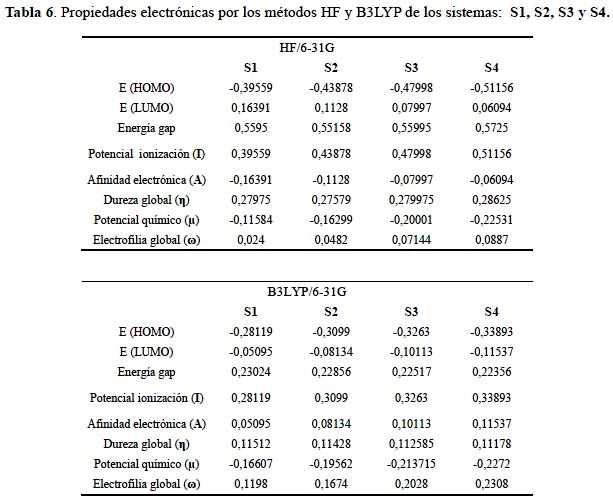
Propiedades electrónicas
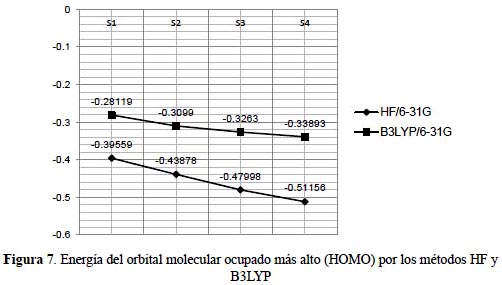
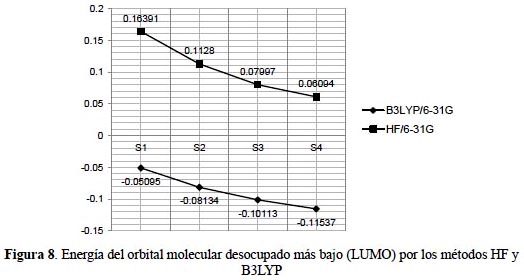
Las propiedades electrónicas en sistemas orgánicos e inorgánicos dependen de los orbitales de frontera HOMO y LUMO. Los orbitales moleculares se extienden entre todos los átomos de una molécula y son combinaciones lineales de orbitales atómicos, los orbitales moleculares que interactúan son generalmente el de más alta energía ocupado HOMO y el de más baja energía desocupado LUMO. La tabla 6 muestra que la brecha de energía HOMO-LUMO denominado energía gap muestra una ligera disminución en relación al incremento de las sustituciones del grupo nitratoxicarbono desde 0,23024 en el sistema S1 a 0,22356 en S4 dentro del contexto del método DFT; en el método de Hartree-Fock sucede de forma contraria; el sistema S1 muestra 0,5595 y el S4 0,5725. La energía gap refleja la estabilidad cinética, la reactividad química, la polarizabilidad molecular y la dureza o blandura14. Los métodos de correlación electrónica mejoran los valores sobre los obtenidos por el método de Hartree-Fock.
El potencial de ionización (I) expresado como I = -EHOMO es superior dentro del método Hartree-Fock con 0,51156 y empleando el funcional B3LYP obtenemos un valor de 0,33893, con una diferencia de 0,17263 en ambos métodos.
La afinidad electrónica (A) expresada como A = -ELUMO muestra valores altos, característico en sistemas que pueden acomodar fácilmente un exceso de electrones provenientes de los alrededores. Los mejores valores de energía del orbital molecular desocupado más bajo LUMO son obtenidos en la teoría DFT.
La dureza química (η) expresado como η = ½(ELUMO ̶EHOMO) es una propiedad global del sistema y mide la resistencia impuesta por éste al cambio en su distribución electrónica. En este contexto la dureza es un descriptor de la reactividad mostrando valores mayores en el método Hartree-Fock.
El potencial químico (µ) expresado como µ = ½(EHOMO ̶ELUMO) es una propiedad donde los electrones tienden a fluir de regiones de baja hacia alta electronegatividad (de regiones de alto potencial químico a regiones de bajo potencial químico). Observamos en la tabla 6 una tendencia que se va acentuando hasta llegar a sustituir todos los hidrógenos por grupos nitratoxicarbono, obteniendo así -0,225 y -0,227 en el método Hartree-Fock y B3LYP respectivamente, siendo el sistema S4 el que presenta el menor potencial químico.
En cuanto a la electrofilia global (ω) expresado como ω = µ2/2η, el sistema S4 entrega un valor de 0,2308 en la teoría del funcional de la densidad siendo el más representativo en esta serie.
CONCLUSIONES
El tetraedrano y sus derivados nitratoxicarbono son teóricamente estudiados a un nivel de cálculo HF/6-31G y B3LYP/6-31G.
El incremento de grupos sustituyentes nitratoxicarbono en el tetraedrano contribuye a la estabilidad de la molécula.
El grupo nitratoxicarbono presenta menor redistribución de carga en los átomos de oxígeno, respecto a los grupos nitro.
AGRADECIMIENTO
Los autores agradecen al Dr. William Tiznado Vásquez de la Universidad Andrés Bello, Santiago de Chile, por brindarnos soporte computacional.
BIBLIOGRAFÍA
1. A. Greenberg & J. F. Liebman, Organic Chemistry, A Series of Monographs, Vol. 38: Strained Organic Molecules, H. H. Wasserman (editor), Academic Press, Inc., U.S.A., 1978, 85-90.
2. G. Maier, Tetrahedrane and Cyclobutadiene. Angew. Chem. Int. Ed. Engl., 1988; 27: 309-332.
3. E. G. Lewars, Modeling Marvels, Computational Anticipation of Novel Molecules, Springer Science+Business Media B.V., 2008, 81-104.
4. B. A. Hess, Jr. & L. J. Schaad, Ab Initio Second-Order Møller-Plesset Calculation of the Vibrational Spectrum of Tetrahedrane. J. Am. Chem. Soc., 1985; 107: 865-866.
5. J. Akhavan, the Chemistry of Explosives (RSC Paperbacks), The Royal Society of Chemistry, 1998, 18-43.
6. G. W. Griffin & A. P. Marchand, Synthesis and Chemistry of Cubanes. Chem. Rev., 1989; 89: 997-1010.
7. P. E. Eaton, R. L. Gilardi & M.-X. Zhang, Polynitrocubanes: Advanced High-Density, High-Energy Materials. Adv. Mater, 2000; 12: 1143-1148.
8. R. M. Richard & D. W. Ball, B3LYP Calculations on the Thermodynamic Properties of a Series of Nitroxycubanes Having the Formula C8H8-x (NO3)x (x= 1-8), Journal of Hazardous Materials. 2009; 164: 1595-1600.
9. D. A. Hrovat, W. T. Borden, P. E. Eaton & B. Kahr, A Computational Study of the Interactions Among the Nitro Groups in Octanitrocubane. J. Am. Chem. Soc., 2001; 123: 1289-1293.
10. A. Nemirowski, H. P. Reisenauer & P. R. Schreiner, Tetrahedrane - Dossier of an Unknown. Chem. Eur. J., 2006; 12: 7411-7420.
11. G. Zhou, J.-L. Zhang, N.-B. Wong & A. Tian, Computational Studies on a Kind of Novel Energetic Materials Tetrahedrane and Nitro Derivatives. Journal of Molecular Structure (Theochem), 2004; 668: 189–195.
12. R. W. Zoellner, C. L. Lazen & K. M. Boehr, A Computational Study of Novel Nitratoxycarbon, Nitritocarbonyl, and Nitrate Compounds and their Potential as High Energy Materials. Computational and Theoretical Chemistry, 2012; 979: 33-37.
13. R. J. Buszek, C. M. Lindsay & J. A. Boatz, Tetrakis (nitratoxycarbon) Methane (Née CLL-1) as a Potential Explosive Ingredient: a Theoretical Study. Propellants Explos. Pyrotech., 2010; 35: 1-5.
14. D. Pegu & N. B. Singh, Quantum Chemical Calculations of Molecular Structure, Electronic, Thermodynamic and Non-linear optical properties of 2-amino-3nitro-6-methyl pyridine. International Journal of Advanced Research, 2013; 1: 531-538.
Recibido el 13-01-2015
Aprobado el 27-01-2015

