










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de la Sociedad Química del Perú
Print version ISSN 1810-634X
Rev. Soc. Quím. Perú vol.82 no.4 Lima Oct./dic. 2016
TRABAJOS ORIGINALES
Calificación de la equivalencia farmacéutica in vitro por la metodología HPLC de cuatro medicamentos conteniendo paracetamol, clorfenamina maleato y fenilefrina clorhidrato en tabletas
Rating pharmaceutical equivalence in vitro by HPLC methodology four drugs containing acetaminophen, chlorpheniramine maleate and phenylephrine hydrochloride tablets
Jhonnel Samaniego Joaquin*a, Gladys Arias Arroyoa
a Facultad Farmacia y Bioquímica, Universidad Nacional Mayor de San Marcos, Jr. Puno 1002. Jardín Botánico. Lima. Perú. jhonnel28@hotmail.com
RESUMEN
Se calificó la equivalencia farmacéutica in vitro de cuatro medicamentos conteniendo paracetamol, clorfenamina maleato y fenilefrina clorhidrato en tabletas utilizando la Cromatografía Líquida de Alta Resolución en fase reversa (HPLC-RP), se determinó los perfiles de disolución del producto de referencia y de los 4 productos en estudio, además se establecieron y compararon las diferencias de los perfiles de disolución del producto de referencia y los productos en estudio. El diseño de la metodología empleada fue analítico, experimental y comparativo. Se evaluaron cuatro formulaciones comercializadas en Lima y el medicamento de referencia mediante el método de cromatografía líquida de alta resolución (HPLC). Se cuantificaron los perfiles de disolución y fueron comparados a través de un método estadístico modelo independiente, factor de diferencia (f1) y de similitud (f2), los cuales revelaron que las curvas de disolución son distintas entre los productos del laboratorio de referencia y los laboratorios analizados, ya que f1 y f2 estuvieron fuera de los rangos establecidos en dos de las cuatro formulaciones evaluadas. Los resultados obtenidos demostraron que dos de los cuatro productos analizados con este método no serían equivalentes farmacéuticos.
Palabras clave: Equivalencia farmacéutica in vitro, cromatografía líquida de alta resolución, perfiles de disolución, factor de diferencia (f1), factor de similitud (f2).
ABSTRACT
Pharmaceutical equivalence in vitro four medicines containing paracetamol, maleate chlorpheniramine and phenylephrine hydrochloride tablets using Chromatography Liquid High Resolution reversed phase (HPLC-RP) is called, the dissolution profiles of the reference product and were determined 4 products in the study also were established and compared the differences in the dissolution profiles of the reference product and the products under study. The design methodology was analytical, experimental and comparative. Four formulations marketed in Lima and the reference product was evaluated by the method of high performance liquid chromatography (HPLC). the dissolution profiles were measured and were compared using a statistical method independent model difference factor (f1) and similarity (f2), which revealed that the dissolution curves are different between products reference laboratory and laboratories analyzed, since f1 and f2 were outside the ranges established in two of the four formulations tested. The results showed that two of the four products tested by this method would not be pharmaceutical equivalents.
Key words: in vitro pharmaceutical equivalence, high-resolution liquid chromatography, dissolution profiles difference factor (f1), similarity factor (f2).
INTRODUCCIÓN
Los estudios de bioequivalencia in vivo han constituido el tipo de evidencia de primera elección para demostrar la intercambiabilidad de la mayoría de los medicamentos. Sin embargo, entidades internacionales como la FDA (Food and Drug Administration) y la EMA (European Medicines Agency) han abierto la puerta a la posibilidad de demostrar la equivalencia in vitro para ciertos medicamentos, mediante ensayos de disolución y pruebas que garantizan su eficacia y seguridad; y así evitar la realización de ensayos in vivo en humanos y disminuir los costos de desarrollo de los medicamentos. Para ello utilizamos las comparaciones de los perfiles de disolución del medicamento en estudio.1
Los medicamentos genéricos son todos aquellos diferentes al innovador. Equivalentes farmacéuticos o alternativas farmacéuticas que pueden o no ser equivalentes terapéuticos. Aquellos que hayan demostrado equivalencia in vivo o in vitro, se consideran terapéuticamente equivalentes al producto de referencia y pueden ser declarados intercambiables. Son equivalentes farmacéuticos aquellos medicamentos que contienen la misma cantidad molar de ingrediente farmacéutico activo, en la misma forma farmacéutica, están destinados a ser administrados por la misma vía y cumplen con estándares de calidad idénticos o comparables.1
Los estudios in vitro están constituidos por estudios comparativos de perfiles de disolución, en donde se determina la cantidad o porcentaje del principio activo disuelta en función del tiempo bajo condiciones controladas y validadas. El propósito de este tipo de estudio es comparar las características de liberación del principio activo contenido en una forma farmacéutica sólida oral de liberación inmediata.2
Las pruebas para realizar los perfiles de disolución deberán realizarse tomando como referencia la metodología establecida en la Farmacopea de los Estados Unidos (USP) en un Aparato I a 100 rpm o un Aparato II a 50 rpm usando 900 mL de los siguientes medios de disolución: (1) 0,1 N de HCl o Fluido Gástrico Simulado USP sin enzimas; (2) un tampón de pH 4,5 y (3) un tampón de pH 6,8 o Fluido Intestinal Simulado USP sin enzimas. Para cápsulas y comprimidos recubiertos de gelatina, se puede usar Fluidos Gástrico o Intestinal Simulado USP (con enzimas). Se deberá evaluar un mínimo de 12 unidades posológicas de un producto medicamentoso para respaldar una solicitud de bioexención. Se deberá recolectar las muestras en un número suficiente de intervalos para caracterizar el perfil de disolución del producto medicamentoso. Cuando se comparan los productos de prueba y referencia, se deberá comparar los perfiles de disolución usando un factor de similitud (f2). El factor de similitud es una transformación logarítmica de la raíz cuadrada recíproca de la suma del error cuadrado y es una medición de la similitud en el porcentaje (%) de disolución entre las dos curvas.3
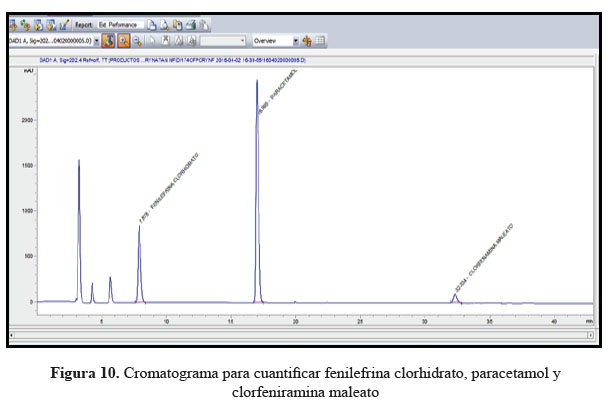
La cuantificación simultánea de las muestras obtenidas de la combinación de paracetamol, clorfenamina maleato y fenilefrina clorhidrato como medicamento antigripal en tabletas, se determinó por el método analítico HPLC ya que es más específico y sensible.4
El objetivo general del presente trabajo fue calificar la equivalencia farmacéutica in vitro de cuatro medicamentos conteniendo paracetamol, clorfenamina maleato y fenilefrina clorhidrato en tabletas utilizando la Cromatografía Líquida de Alta Resolución en fase reversa (HPLC-RP).
Los objetivos específicos fueron la determinación de los perfiles de disolución del producto de referencia y de los 4 productos en estudio, además comparar las diferencias de los perfiles de disolución del producto de referencia y los productos en estudio.
PARTE EXPERIMENTAL
Reactivos
Ácido clorhídrico (Merck), fosfato monobásico de potasio (Merck), acetonitrilo (grado HPLC, J.T. Baker), ácido fosfórico al 85% (calidad R.A. J.T. Baker), agua (HPLC).
Material de referencia
Clorfeniramina maleato (estándar primario USP), Paracetamol (estándar primario USP) y Fenilefrina clorhidrato (estándar primario USP),
Equipos
• Sistema HPLC Agilent Tecnologies 1260, equipado con bomba 1260-G1311B, auto muestreador 1260 ALS-G1329B, horno para columna l260 TCC-G1316A y detector de arreglo de diodos 1260 DAD-G4212B. Para la adquisición y procesamiento de los cromatogramas, se empleó el programa Open lab. • Equipo de disolución Sotax AT7 Smart • Balanza analítica Mettler Toledo XPE205
Sistema cromatográfico
Se empleó como fase móvil una solución fosfato monobásico de potasio, se ajustó a pH 3,5 con ácido fosfórico al 85%. Se empleó una columna L11 relleno de grupos fenilo de sílice de 150 x 4,6 mm 5 μm (Agilent). Se trabajó a un flujo de 0,5 mL/min hasta el minuto 7,0 y luego se aumentó el flujo a 1,0 mL/min hasta el minuto 22,0 de forma constante hasta el minuto 35,0 y luego se disminuye el flujo a 0,5 mL/min hasta el minuto 40,0. También cuenta con una elución en gradiente de acetonitrilo a los 22,0 minutos en un 20% hasta el minuto 35,0 y luego continuo con fase móvil. Se empleó detección UV a 205 ŋm para Clorfeniramina maleato, 202 ŋm para Fenilefrina clorhidrato, 298 ŋm para Paracetamol; temperatura de la columna 30ºC; volumen de inyección: 20μL.
Método
Se utilizó el método de cromatografía líquida de alta resolución en fase reversa (HPLC-RP) en fase reversa para determinar la equivalencia in vitro de la combinación de paracetamol, clorfenamina maleato y fenilefrina clorhidrato tabletas.4
Tipo y diseño de investigación
Estudio analítico experimental y comparativo, empleándose cuatro formulaciones de la combinación de paracetamol, clorfenamina maleato y fenilefrina clorhidrato tabletas comercializados en la ciudad de Lima, en establecimientos farmacéuticos los cuales serán comparados con el original de laboratorio multinacional (medicamento de referencia).
Unidad de análisis
Se procedió a validar un método de ensayo por HPLC para determinar la equivalencia in vitro de la combinación de paracetamol, clorfenamina maleato y fenilefrina clorhidrato tabletas.
Tamaño de muestra
Cinco en total: cuatro formulaciones comercializadas en Lima y el medicamento de referencia. Para calificar la equivalencia farmacéutica in vitro se comparó simultáneamente cada uno de los medicamentos en estudio con el producto de referencia.
Selección de muestra
Se procedió a obtener las muestras de las cuatro formulaciones, adquiriéndolas en cuatro cadenas de boticas y farmacias de mayor influencia en el mercado farmacéutico que son comercializadas en la ciudad de Lima.
Técnica de recolección de datos
Los perfiles de disolución fueron cuantificadas a través de la técnica de cromatografía líquida de alta resolución en fase reversa (HPLC-RP). Estos perfiles se compararon a través de un método estadístico modelo independiente, factor de diferencia (f1) y de similitud (f2).3
Determinación de los perfiles de disolución:
• Preparación de la solución estándar
Se pesaron 46,5 mg de Fenilefrina clorhidrato y 18,5 mg de Clorfeniramina maleato, se transfirieron a un matraz volumétrico de 250 mL. Se añadieron 100 mL fase móvil y se sónico por 5 minutos y se enrasó con fase móvil. (Fiola A). Se pesaron 55,0 mg de paracetamol y se transfirieron a un matraz volumétrico de 100 mL. Se añadieron 50 mL de fase móvil y se sónico por 5 minutos. Se transfirió 3 mL de la Fiola A y se enrasó con fase móvil. Se homogeneizó y filtró por membrana de 0,45μm PVDF Millipore. Concentración aproximada de 0,0056 mg/mL de fenilefrina clorhidrato, 0,0022 mg/mL de clorfeniramina maleato y 0,55 mg/mL de paracetamol.
• Preparación de las muestras
Se evaluaron doce tabletas de cada formulación por lote y en todos los procesos, de igual concentración nominal de 5 mg de fenilefrina clorhidrato, 5 mg de clorfenamina maleato y 500 mg de paracetamol para todas las muestras. El volumen de medio de disolución a emplear fue de 900 mL al que se le agrego las formulaciones en estudio y se muestrearon de manera manual y con 5 tiempos de muestreo recomendados por la FDA establecidos a los 15, 30, 45, 60 y 75 minutos. Los perfiles de disolución se realizaron usando aparato 2 USP a 50 rpm empleando equipo de disolución ZOTAX. Los medios de disolución fueron soluciones amortiguadoras USP (11) para simular los fluidos fisiológicos del sistema gastrointestinal: pH 1,2 (fluido gástrico simulado sin enzimas), pH 4,5 (solución buffer acetato), y pH 6,8 (solución buffer fosfato) a una temperatura de 37 ± 0,5ºC. Todas las muestras se filtraron a través de membranas de 0,45 μm PVDF Millipore. Concentración aproximada de 0,0056 mg/mL de fenilefrina clorhidrato, 0,0056 mg/mL de clorfeniramina maleato y 0,55 mg/mL de paracetamol.
RESULTADOS Y DISCUSIÓN
a. Comparación de los perfiles de disolución para las cuatro formulaciones en estudio y el producto de referencia en medio de disolución a pH 1,2
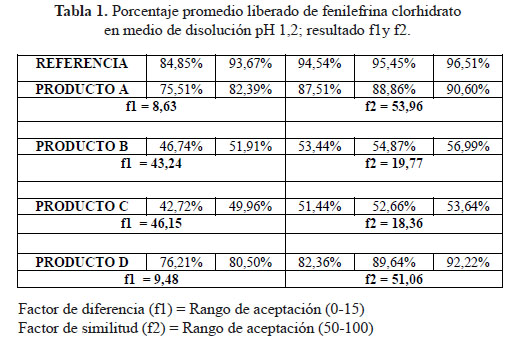
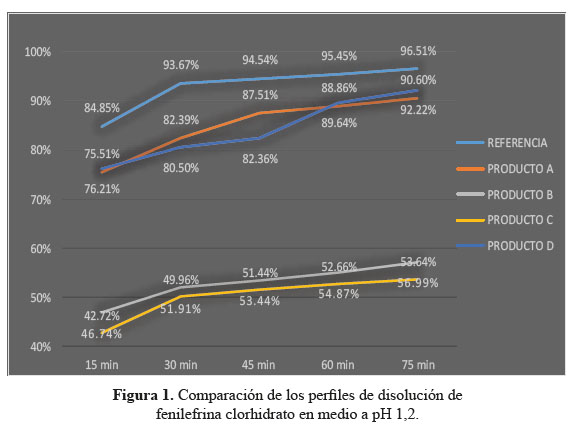
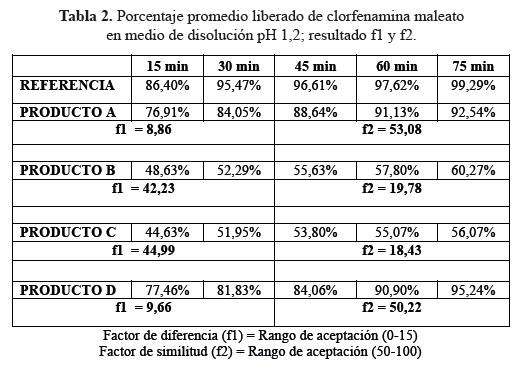
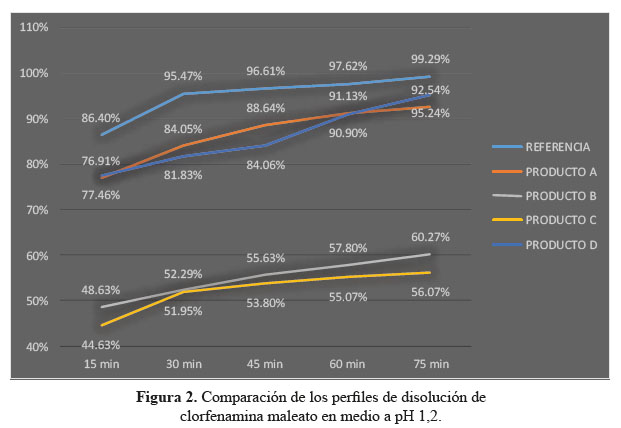
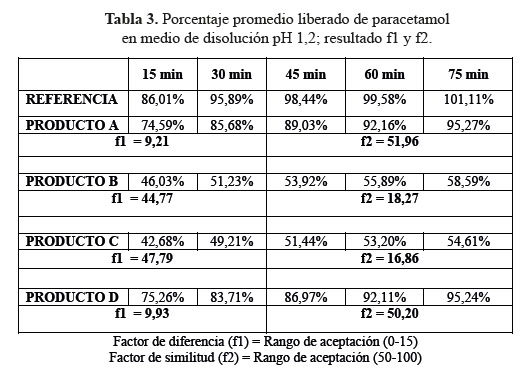
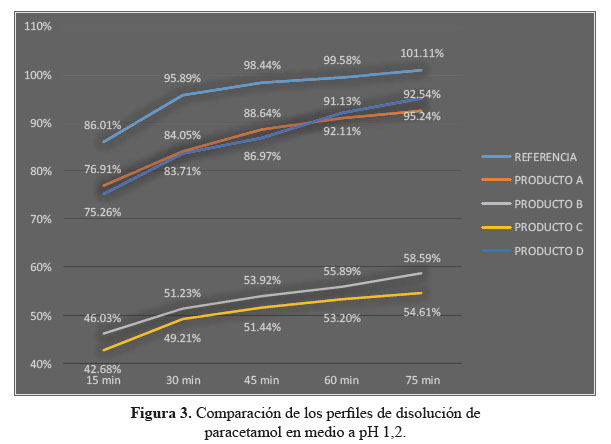
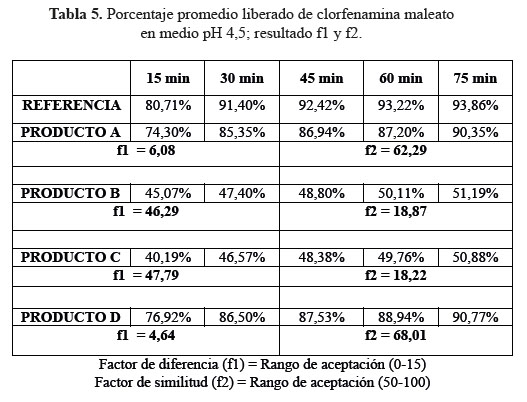
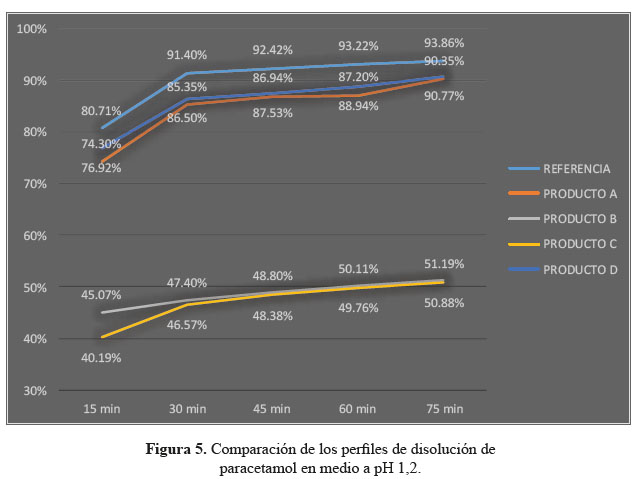
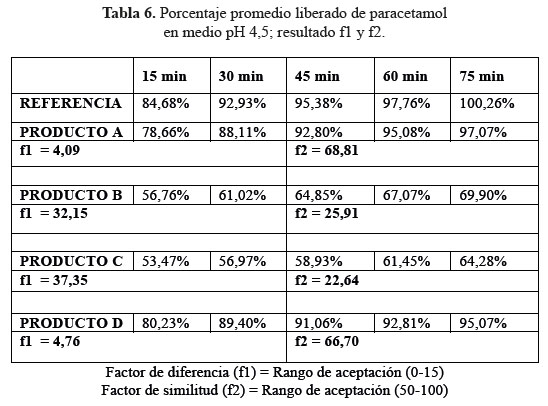
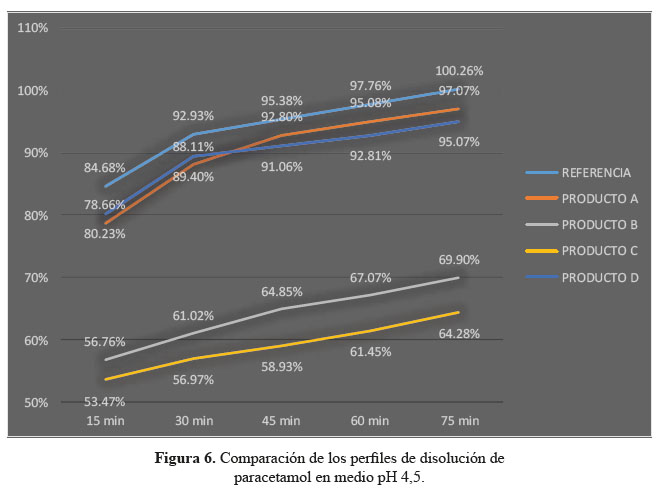
b. Comparación de los perfiles de disolución para las cuatro formulaciones en estudio y el producto de referencia en medio de disolución a pH 4,5.
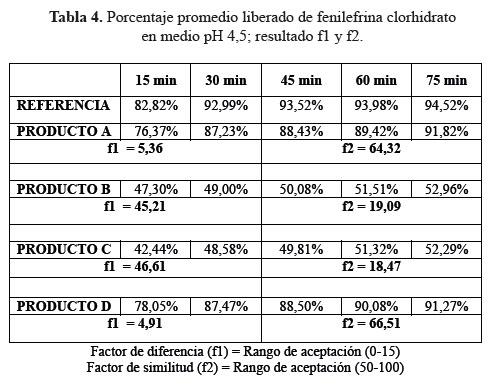
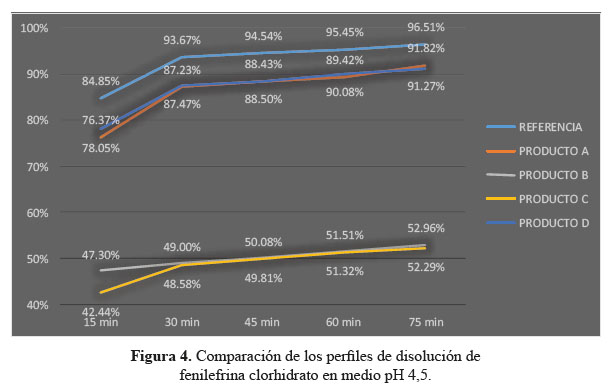
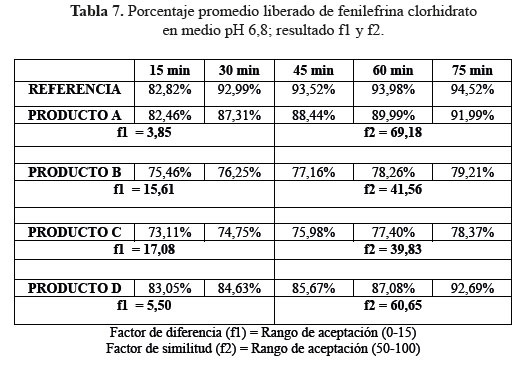
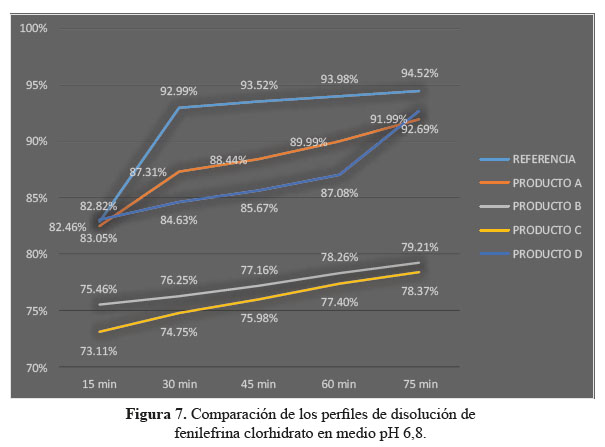
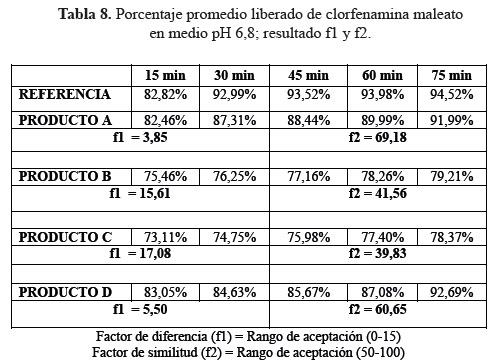
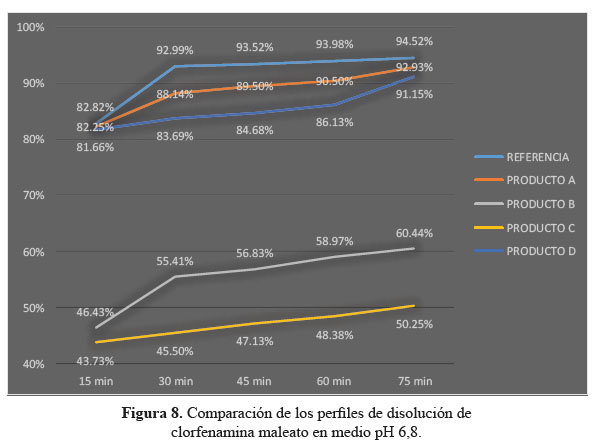
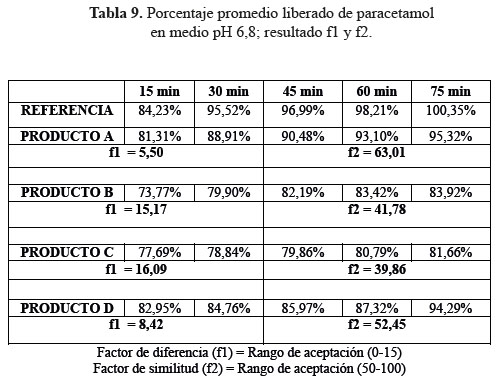
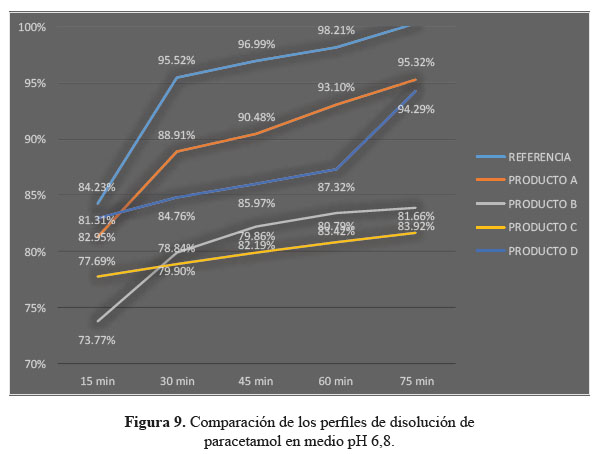
c. Comparación de los perfiles de disolución para las cuatro formulaciones en estudio y el producto de referencia en medio de disolución a pH 6,8.
DISCUSIÓN
En el desarrollo de los perfiles de disolución según las tablas 1, 2 y 3, las formulaciones B y C, evidenciaron una baja disolución en medio de disolución a pH 1,2 en comparación con las formulaciones A y D también evaluadas las cuales sí resultaron ser similares al compararlas con el producto de referencia, según los datos obtenidos como el factor de similitud (f2) y factor de diferencia (f1).3
En las tablas 4, 5 y 6, las formulaciones A y D, logaron una mejor disolución en medio de disolución a pH 4,5 obteniendo factores de similitud (f2) y factores de diferencia (f1) aceptables, en comparación a las formulaciones B y C que no alcanzaron el mínimo requerido para ambos factores; determinando según este modelo que no son similares al producto de referencia.3
Las determinaciones de los perfiles de disolución de las formulaciones en estudio C y D en medio de disolución a pH 6,8, según tablas 7, 8 y 9, no lograron establecer su similitud con respecto al producto de referencia según la evaluación de los factores de similitud (f2) y factores de diferencia (f1); con respecto a las formulaciones A y D sí se obtuvieron valores aceptables, lográndose determinar su similitud según el análisis de los factores antes mencionados.3
Se observaron diferencias con respecto a la liberación de los principios activos entre el producto de referencia que liberó más del 80% a los 15 minutos en los tres diferentes medios a pH 1,2, 4,5 y 6,8 y las formulaciones en estudio que no tuvieron el mismo comportamiento logrando liberar en promedio más del 80% solo a los 30 minutos. Esta variación puede deberse a los diferentes excipientes empleados en la formulación de las diversas tabletas y el producto de referencia.8
CONCLUSIONES
Se utilizó una técnica analítica por Cromatografía Líquida de Alta Resolución en fase reversa (HPLC-RP) para lograr determinar la equivalencia in vitro de la combinación de paracetamol, clorfenamina maleato y fenilefrina clorhidrato en tabletas; ya que presenta mayores ventajas por ser el principal método de separación de especies químicas estrechamente relacionadas.
Se logró desarrollar los perfiles de disolución de los de las cuatro formulaciones y el producto de referencia, los cuales fueron evaluados de acuerdo a un método modelo independiente f1 y f2. Estos revelaron que dos formulaciones, B y C, no son equivalentes in vitro, ya que tienen un comportamiento diferente durante el desarrollo de los perfiles de disolución.
REFERENCIAS BIBLIOGRÁFICAS
1. Ministerio de Salud Dirección General de Medicamentos Insumos y Drogas. Directiva Sanitaria que regula los estudios de Equivalencia Terapéutica para demostrar la intercambiabilidad de medicamentos. Lima, 2014 [ Links ]
2. Medina A. Bioexenciones y estudios de bioequivalencia in vitro. [en línea] Revista electrónica del Centro de la Ciencia y la Investigación Farmacéutica. Colombia. 2009,5: 9-15. [accesado Febrero del 2009]. Disponible en: http://www.clinicaces.com/userfiles// Magazine-CECIF-No%20-5-Feb-Mar-2009.pdf [ Links ]
3. U.S. Food And Drug Administration. Exención de los estudios de biodisponibilidad y bioequivalencia in vivo para formas posológicas orales sólidas de liberación inmediata en base a un sistema de clasificación de biofarmacéuticas. Guía para la Industria. Rockville: Food and Drug Administration; 2000. [ Links ]
4. Samaniego J, Arias G. Desarrollo y validación de una metodología analítica por HPLC para la cuantificación simultánea de fenilefrina clorhidrato, paracetamol, salicilamida, cafeína y clorfenamina maleato en tabletas. Rev Soc Quim Perú. 2016; 82.(2):196-207 [ Links ]
5. Farmacopea de los Estados Unidos de América. USP 38 NF 33. Maryland: The United States Pharmacopeia Convention; 2015. [ Links ]
6. Herrera O, Grande M. Equivalencia terapéutica de tabletas de diazepam dispensadas en la ciudad de Ica, Perú. Rev Med Hered. 2012; 23(3):154-159. [ Links ]
7. Villalba O, Ortiz J, Grande M, Isasi J, Yantas D, Fiestas V. Estudio de Bioequivalencia de Ibuprofeno 400 mg. Tabletas. Rev Perú Med Exp Salud Pública. 2007; 24 (4):356- 362. [ Links ]
8. European Medicines Agency (EMA). Committee for medicinal products for human use. Guideline on the Investigation of Bioequivalence. London: European Medicines Agency; 2010. [ Links ]
Recibido el 29.08.2016
Aprobado el 22.12.2016
