











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La estrategia farmacológica llamada efecto caballo de Troya (THA, por sus siglas en inglés: Trojan Horse Approach)) alude a la Guerra de Troya para explicar cómo se promueve la inserción de un agente tóxico (fármaco) en bacterias, empleando su propio mecanismo bioquímico de adquisición de hierro. En nuestra atmósfera oxidante predomina el Fe3+, sin embargo, este presenta una extremadamente baja biodisponibilidad1. Las bacterias y otros microorganismos desarrollaron estrategias de quelación para favorecer la formación de complejos de alta estabilidad con Fe3+, para ello, sintetizan moléculas quelantes de bajo peso molecular llamadas sideróforos, como por ejemplo, enterobactina en Escherichia coli o preacinetobactina en Acinetobacter baumannii2. El complejo Fe3+-sideróforo se puede acoplar en una proteína receptora de sideróforos (endógenos y/o exógenos) localizada en la membrana externa del microorganismo, mediante un conjunto de interacciones intermoleculares con algunos residuos de aminoácidos específicos en la cavidad del receptor que inducen un ajuste dinámico entre el ligando y el receptor3. Esta configuración de interacciones, permite el reconocimiento molecular y promueve el proceso de internalización del hierro- sideróforo4.
A. baumannii es un cocobacilo Gram-negativo oportunista con alta supervivencia y resistencia a los antibióticos, en comparación a otras especies de Acinetobacter no baumannii. Se asocia principalmente con infecciones nosocomiales en pacientes de cuidados intensivos e inmunocomprometidos. Lamentablemente, la falta de tratamientos eficaces resulta en altas tasas de mortalidad, por ello, la Organización Mundial de la Salud (OMS) considera a A. baumannii resistente a carbapenémicos como una prioridad 1 o crítica, que requiere nuevos antibióticos con urgencia5,6.
En nuestro organismo, el mecanismo de defensa promovido por citoquinas, hormonas y proteínas como las lipocalinas7,8 restringen la disponibilidad de hierro a concentraciones extremadamente bajas (hasta 10−24 M), en consecuencia, A. baumannii, expresa receptores de xenosideróforos tales como desferricoprogen, ácido rodotorúlico y desferrioxamina para incrementar sus posibilidades de captación de hierro9. Los sideróforos endógenos identificados en A. baumannii son de tipo catecol y son la acinetobactina, la fimsbactina y la baumannoferrina, que son transportadas por diferentes tipos de proteínas receptoras, lo cual destaca su especificidad. En condiciones de alta demanda de hierro, participan proteínas receptoras de sideróforos de otros tipos, como reportaron Grinter y Lithgow en el año 2019 10, al cristalizar por primera vez el hierro- sideróforo hidroxámico coprogeno (HWS) en la proteína FhuE (por sus siglas en inglés ferric hydroxamate uptake E), identificado con 6E4V en el repositorio de proteínas PDB RSCB.

La Desferrioxamina B (DFO, Figura 1) o N-(5-aminopentil)-N-hidroxi-N'-[5-(N-hidroxi- 3-{[5-(N-hidroxiacetamido)pentil]carbamoilo}propanamido)pentil]butanodia-mida (C25H48N6O8), es secretada por Streptomyces pilosus, y puede ser reconocida por otros microrganismos con receptores de sideróforos de tipo hidroxámico, como es el caso de la proteína FhuE. DFO presenta dos constantes de acidez, 7.92 en los grupos hidroxámicos y 10.23 en el grupo amina, que en medio neutro forma una especie catiónica. La carga total +1 se mantiene al coordinarse con hierro y formar el compuesto Ferrioxamina (Fe- DFO). Se han reportado casos de éxito de aplicación de THA en algunas bacterias Gram negativas, empleando conjugados de DFO con fármacos o, por otro lado, complejos de DFO coordinando con metales abiogénicos, como Cd2+, Al3+ (Al-DFO)11 o Ga3+ (Ga- DFO)12, sin embargo, todavía no se estudiaron contra A. baumannii.
Al3+ y Ga3+, son parecidos con el ion férrico en el radio iónico (dependiendo de su spin), radio covalente, producto de solubilidad de Me(OH)3 (donde Me se refiere al metal) y constante de estabilidad al coordinarse con DFO para formar MeIII-DFO (Tabla 1), sin embargo, difieren en configuración electrónica, estructura magnética, temperatura de fusión, densidad y potencial de reducción. En consecuencia, el hierro presenta una marcada diferencia en su abundancia en el universo, en la corteza terrestre y a diferencia de Al y Ga, es esencial su participación en reacciones bioquímicas.
Tabla 1 Propiedades de los elementos aluminio, galio y hierro.
| Aluminio | Galio | Hierro | |
| Número atómico | 13 | 31 | 26 |
| Abundancia (%) | |||
| Universo | 0,005 | 1x10-6 | 0,11 |
| Corteza terrestre | 8,1 | 0,0019 | 6,3 |
| Humanos | - | - | 0,006 |
| Configuración electrónica | |||
| Neutro | [Ne] 3s2 3p1 | [Ar] 3d10 4s2 4p1 | [Ar] 3d6 4s2 |
| Estado de oxidación 3+ | [Ne] | [Ar] 3d10 | [Ar] 3d5 |
| Electronegatividad de | 1,61 | 1,81 | 1,83 |
| Pauling | |||
| I1 / I2 / I3 (kJ mol-1) | 577 / 1816 / 2744 | 579 / 1979 / 2963 | 762 / 1562 / 2957 |
| Conductividad eléctrica | 3,8x107 | 7,1x106 | 1,0x107 |
| (S/m) | (conductor) | (conductor) | (conductor) |
| Tipo magnético | Paramagnético | Diamagnético | Ferromagnético |
| Temperatura de fusión (°C) | 660,32 | 29,76 | 1538 |
| Densidad (g/cm3) | 2,7 | 5,9 | 7,8 |
| Potencial de electrodo (V) | -1,66 | -0,549 | 0,77 |
| {E°, Me3+ + 3e- → Me} | (Fe3+/Fe2+) | ||
| Radio iónico trivalente | 0,54 | 0,62 | 0,65 d5 spin alto |
| (Å), NC=6 | 0,55 d5 spin bajo | ||
| Radio covalente en Me3+ (Å) | 1,18 | 1,26 | 1,25 |
| pKa1 del [Me(H2O)6]3+ | 4,99 | 2,6 | 2,2 |
| Log Kps [Me(OH)3] | -33,5 | -37 | -38 |
| Log β [MeIII-DFO] | 36,11 | 38,96 | 42,33 |
De los estudios in vitro realizados empleando Al, Ga y Fe con DFO (Me-DFO), principalmente se apoyaron de las concentraciones inhibitorias para validar la aplicación de THA, y no se realizó un análisis de las interacciones ligando-receptor. En este trabajo se estudiaron las similitudes y diferencias en reactividad química entre Al-DFO, Ga-DFO y Fe-DFO, y se analizaron sus interacciones intermoleculares con FhuE, empleando cálculo mecánico cuántico, docking molecular y dinámica molecular durante 50 ns, para mejorar la comprensión de los sistemas metal-DFO-FhuE.
PARTE COMPUTACIONAL
Optimización estructural y reactividad química
Las coordenadas cartesianas de los átomos de Fe-DFO se obtuvieron de la base de datos cristalográficos de Cambridge (CSD) con el identificador 155586. Se eliminaron las aguas de cristalización y los contraiones. Se usó Gaussian0913 para optimizar la estructura molecular de Fe-DFO, empleando la Teoría del Funcional de la Densidad (DFT en sus siglas en inglés) al nivel B3LYP14,15/6-31G(d,p)16,17 para los átomos de C, H, O y N, la base SDD18 adaptada al respectivo pseudopotencial para el átomo metálico. Las correcciones del efecto del solvente han sido consideradas usando el modelo de solvatación implícita SMD19. Los estados estacionários de mínimo no presentaron ninguna frecuencia imaginaria. Se usó el programa Schrödinger20, para calcular la desviación cuadrática media de las estructuras superpuestas. Los cálculos con Gaussian09 fueron realizados en el clúster Heimdall del Grupo de Química Computacional Aplicada de la Universidade de São Paulo (Brasil).
Se empleó la estructura de Fe-DFO como plantilla para generar las estructuras de Al-DFO y Ga-DFO. Estas se optimizaron con el mismo nivel de cálculo mencionado y se verificó la ausencia de frecuencias imaginarias. Empleando el Teorema de Koopmans se calcularon los indicadores de reactividad global de Me-DFO (Me: Fe3+, Al3+ y Ga3+). Para el tratamiento de datos y la generación de gráficas usamos Jmol v1421, Matplotlib 3.2.222, Maestro 202320 y Chemcraft v1.823.
Acoplamiento molecular de Me-DFO y FhuE
Empleamos las estructuras optimizadas de Me-DFO y la proteína FhuE, disponible en el banco de datos de proteínas (PDB RCSB) con identificador 6E4V. FhuE recibió un pretratamiento con Wizard de la suite Schrödinger, donde se eliminaron aguas de cristalización y el ligando co-cristalizado HWS (Ferric coprogen, C35H53FeN6O13). Se editó el estado de protonación del sistema según el pH 7.4 con PropKa y se minimizó la energía del sistema empleando mecánica molecular con el campo de fuerza OPLS. Para la configuración del acoplamiento molecular, se generó el grid box empleando HWS como centro de la caja cúbica con 20 Å de arista. El acoplamiento molecular fue realizado con Glide de la suite Schrödinger en modo extra precisión y el análisis de interacciones en dos dimensiones fue realizado con LigPlot+24.
Simulaciones de dinámica molecular
Empleamos las estructuras acopladas en la etapa anterior para preparar los sistemas de dinámica molecular para cada metal-DFO-FhuE. La caja de simulación consistió en una caja ortorrómbica con aristas que distan 10 Å de la parte más externa de la proteína, se adicionaron moléculas de agua usando TIP3P como modelo de solvente, iones de sodio y cloro para neutralizar el sistema y se estableció una concentración salina de 150mM. Con el campo de fuerza OPLS3, se equilibró el sistema usando Desmond25 con los ensambles isotérmico-isocórico e isobárico-isotérmico, a temperatura 310K con el termostáto de Nosé-Hoover y una presión de 1.01325 bar con el baróstato de Martyna- Tobias-Klein, por 10 y 20 ps, respectivamente. Con este sistema equilibrado se realizó la etapa de producción de 50 ns para cada metal-DFO-FhuE, manteniendo constante el número de partículas, la temperatura (310K) y la presión (1.0 bar) (NPT), con la configuración predeterminada de relajación antes de la simulación26.
La animación de la trayectoria fue generada con 1000 frames por sistema y se disponibilizó en nuestro repositorio de Github. Se usó el módulo SID (Simulation interactions diagram) para la generación de la desviación cuadrática media (RMSD), fluctuación cuadrática media de la proteína (RMSF) y del ligando (L-RMSF), diagrama de interacciones intermoleculares y porcentajes, diagrama de barras de la fracción de interacciones, diagrama de contactos ligando-proteína y propiedades del ligando (Ligand- RMSD, radio de giro, interacciones intramoleculares, área de superficie molecular, área de la superficie accesible al solvente y área de la superficie polar). Los cálculos con el programa Schrödinger fueron realizados en LIBIPMET, de la Universidad Nacional de Ingeniería.
RESULTADOS Y DISCUSIÓN
Optimización estructural y reactividad química
Se obtuvo un RMSD de 0.8 Å entre la estructura cristalizada y la optimizada de Fe-DFO (Figura 2). Esto nos permitió validar el nivel cálculo para usarlo posteriormente en las moléculas de Al-DFO y Ga-DFO.
Las distancias promedio de Fe3+-O(C), Fe3+-O(N) y N-C(hidroxamato) fueron de 2.12,
2.00 y 1.33 Å respectivamente, con similitud a lo reportado en la literatura27,28. Las frecuencias promedio de estiramiento de los grupos amida e hidroxamato fueron 1678 y 1505 cm-1, respectivamente, que son valores aproximados a los reportados por Edwards y colaboradores29. Se observan magnitudes análogas en las cargas de Mulliken de casi todos los átomos, siendo más marcada la diferencia en los átomos de N y O en el hidroxamato (átomos 1, 2, 5, 6, 9 y 10 de la Figura 3) y principalmente, en los átomos metálicos, donde observamos 1.6, 1.2 y 0.7 para Al, Ga y Fe, respectivamente. El vector momento dipolar de los complejos no es paralelo, sin embargo, presenta una direccionalidad aproximada al grupo amina y un módulo mayor en Al-DFO, que en Ga- DFO y Fe-DFO, que son más cercanos en magnitud (Figura 4).
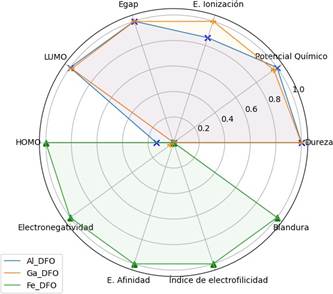
Fe-DFO, presentó una menor barrera energética entre sus orbitales frontera HOMO y LUMO (Egap) en comparación con los otros complejos (Tabla 2). Aunque los orbitales HOMO, son análogos en magnitud, LUMO es -2.47 eV para el Fe-DFO mientras que para Al-DFO y Ga-DFO es positivo, con 0.13 y 0.09, respectivamente. De acuerdo con el Teorema de Koopmans, encontramos que la reactividad química de los complejos es muy distinta (Figura 5). En comparación con los complejos de aluminio y galio, encontramos que Fe-DFO presenta una menor energía de ionización, mayor potencial químico electrónico, mayor energía de afinidad, menor dureza, mayor blandura y mayor índice de electrofilicidad.
Este mayor grado de polarizabilidad y afinidad por regiones apolares, es favorable en el reconocimiento molecular de Fe-DFO, ya que las cavidades de interacción son predominantemente hidrofóbicas en las proteínas receptoras de membrana, como por ejemplo, en FoxA de Pseudomonas aeruginosa 4 o en FhuA de Escherichia coli30.
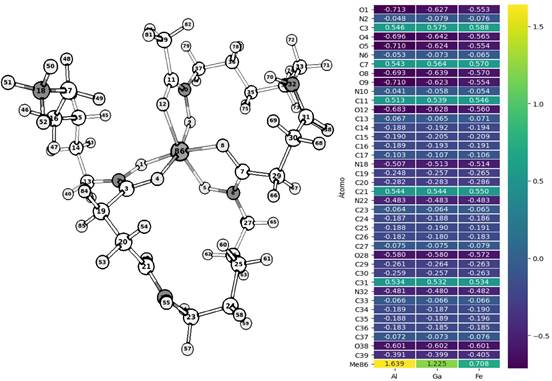
Figura 3 Cargas de Mulliken de los átomos en Me-DFO (Me=Al3+, Ga3+ o Fe3+). Se suprimieron los hidrógenos para mejorar la visualización.
Tabla 2 Reactividad global de los complejos Me-DFO.
| Compuesto | HOMO / eV | LUMO / eV | Energía gap / eV | Energía de ionización | Afinidad Electrónica |
| Al-DFO | -5.83 | 0.13 | 5.96 | 5.83 | -0.13 |
| Ga-DFO | -5.87 | 0.09 | 5.96 | 5.87 | -0.09 |
| Fe-DFO | -5.62 | -2.47 | 3.15 | 5.62 | 2.47 |
Continuación de la Tabla 2.
| Compuesto | Potencial químico | Electronegatividad | Dureza | Blandura | Índice de electrofilicidad |
| Al-DFO | -2.85 | 2.85 | 2.98 | 0.17 | 1.37 |
| Ga-DFO | -2.89 | 2.89 | 2.98 | 0.17 | 1.4 |
| Fe-DFO | -4.04 | 4.04 | 1.57 | 0.32 | 5.19 |
Acoplamiento molecular de Me-DFO y FhuE
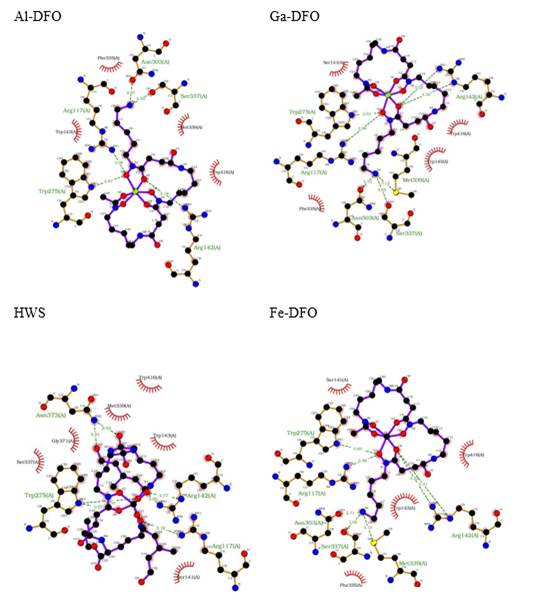
Aunque hay una marcada diferencia en la reactividad química de los iones metálicos en estudio y también de sus complejos con DFO, estas propiedades parecen no influir en el reconocimiento molecular en el receptor FhuE, ya que en las poses de acoplamiento con mayor afinidad entre Me-DFO y FhuE, encontramos ligeras diferencias energéticas y estructurales. Como, por ejemplo, 5.880 kcal/mol de Fe-DFO frente a -5.772 y -5.926 kcal/mol para Al-DFO y GaDFO, respectivamente (Tabla 3). Y en cuanto a la estructura ligando-receptor (Figura 6), se tienen prácticamente los mismos aminoácidos interactuando, incluso considerando al coprogeno (HWS) cocristalizado en FhuE: Arg117, Arg142, Trp275, Asn303, Ser337 y Met339 por enlaces de hidrógeno; y Trp143 y Trp416, por interacciones lipofílicas.
Tabla 3 Docking score e interacciones intermoleculares de Me-DFO con FhuE.
| Compuesto | Docking score / kcal.mol-1 | Enlace de hidrógeno | Lipofílico |
| HWS | -5.814 | Arg117, Arg142, Trp275, Asn373 | Ser141, Trp143, Ser337, Met339, Gly371, Trp416 |
| Al-DFO | -5.772 | Arg117, Arg142, Trp275, Asn303, Ser337 | Trp143, Phe335, Met339, Trp416 |
| Ga-DFO | -5.926 | Arg117, Arg142, Trp275, Asn303, Ser337, Met339, | Ser141, Trp143, Phe335, Trp416 |
| Fe-DFO | -5.880 | Arg117, Arg142, Trp275, Asn303, Ser337, Met339 | Ser141, Trp143, Phe335, Trp416 |
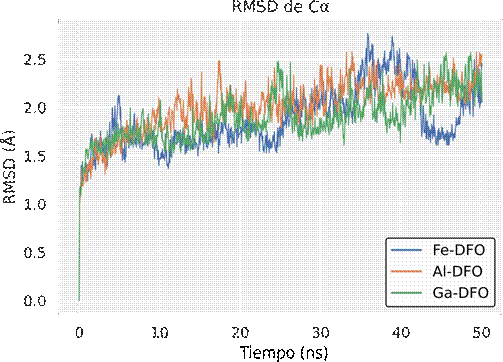
Simulaciones de dinámica molecular
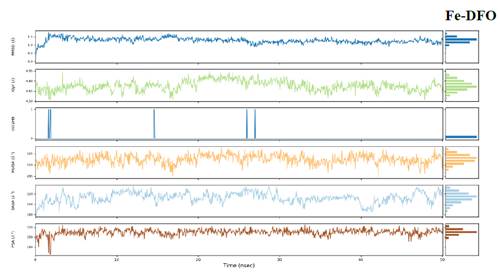
La Figura 7 muestra que los carbonos α en el receptor evolucionan hacia 2.0-2.5 Å de RMSD en los 3 casos. No se observan grandes diferencias entre los sistemas evaluados.
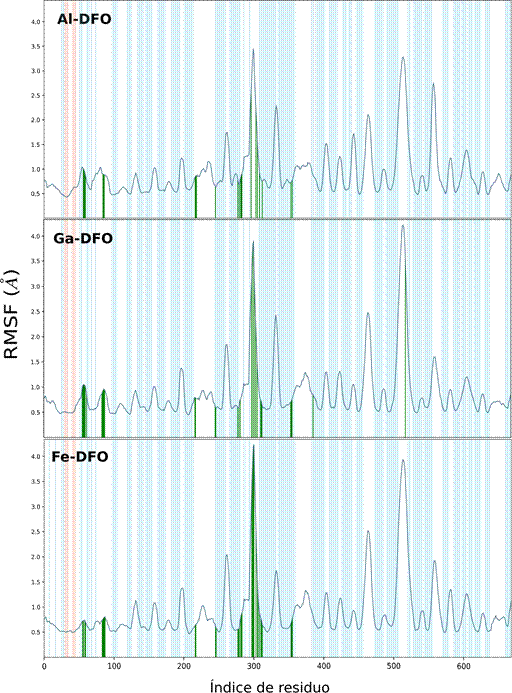
La desviación cuadrática media de las fluctuaciones de los residuos (RMSF) permitieron reconocer un perfil análogo entre Me-DFO (Figura 8), y coincidencias en los puntos de contacto (línea vertical de color verde). No se observaron cambios conformacionales (celeste: hoja beta y anaranjado: alfa) durante los 50 ns.
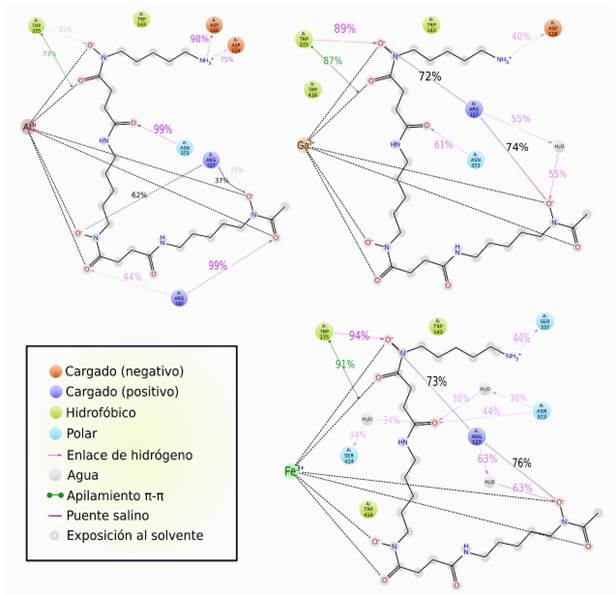
Identificamos interacciones intermoleculares destacadas durante la simulación. La Figura 9, muestra únicamente las mayores al 30%. A continuación, se analizan las interacciones con aminoácidos que se han conservado o no de acuerdo con la información proveniente del docking molecular, empleando las Figuras 9 y 10.
Interacciones intermoleculares recurrentes conservadas:
Arg117: Se ha mantenido con interacción de tipo iónica, debido a su naturaleza catiónica, dirigida hacia los átomos negativos en los hidroxamatos y también mediante enlace de hidrógeno directamente a un oxígeno hidroxámico en Al-DFO, mientras que en Ga-DFO y Fe-DFO, a través de una molécula de agua como intermediaria (puente de agua).
Arg142: Altamente relevante en Al-DFO, mediante enlace de hidrógeno con el oxígeno neutro de dos hidroxamatos, en 44 y 99% (Figura 9). En Ga-DFO y Fe-DFO no hay enlace de hidrógeno, encontramos interacciones menores a 30% empleando aguas como intermediarios, como se observa en color azul en la Figura 10.
Trp143: Interacciones lipofílicas en los tres casos (Me-DFO), incluso también en HWS. Trp275: En Al-DFO ocurre apilamiento π en un 73% y enlace de hidrógeno en 31%. Esto se intensifica en Ga-DFO: 87 y 89%; y Fe-DFO con 91 y 94%, respectivamente.
Trp416: No aparece en Al-DFO, pero sí en los complejos de Ga y Fe, mediante interacciones hidrofóbicas.
Interacciones intermoleculares recurrentes no conservadas:
Ser141, Asn303, Phe335, Ser337 y Met339 aparecen en el acoplamiento en la Tabla 3 de Me-DFO, sin embargo, no durante la dinámica molecular.
Otras Interacciones intermoleculares recurrentes:
Asn373: Mediante enlace de hidrógeno, con 99% en Al-DFO, 61% en Ga-DFO y 44% en Fe-DFO, dependiente de una molécula de agua en los complejos de Ga y Fe.
Asp114: Enlace de hidrógeno con la amina de Al-DFO (75%).
Asp116: Con Al-DFO (98%) y Ga-DFO (40%) mediante enlace de hidrógeno.
En la Figura 10, en general, observamos un perfil de la fracción de interacciones más parecido entre Ga-DFO y Fe-DFO. En Al-DFO se observa como principal aporte el enlace de hidrógeno (barras verdes).
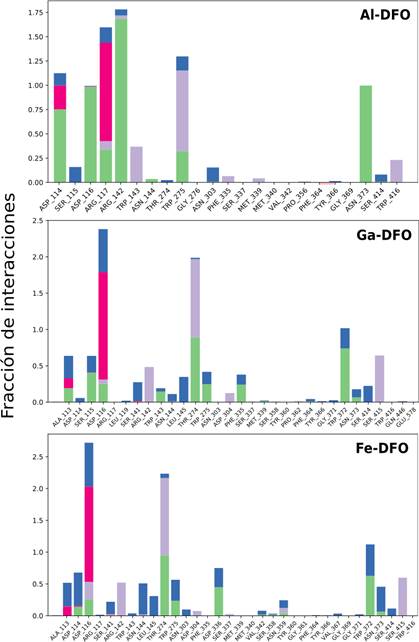
Figura 10 Diagrama de barras de la fracción de interacciones. Paleta de colores usadas en las interacciones: enlace de hidrógeno (verde), puente de agua (azul), iónicas (rojo) e hidrofóbicas (morado).
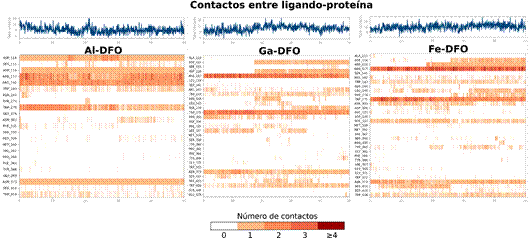
La Figura 11 muestra los momentos (o frames) en que ocurren las interacciones y en asociación con el número de contactos entre Me-DFO y la cavidad de FhuE, se observa como Al-DFO presenta un perfil más distante respecto de Ga-DFO y Fe-DFO.
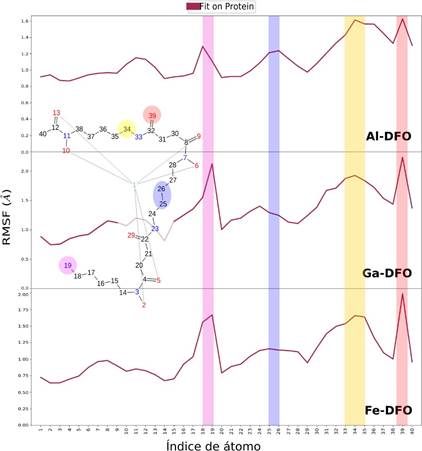
La Figura 12 muestra que los átomos 19, 25, 26 y 39 en Me-DFO tienen mayor movilidad durante la dinámica molecular, mientras que el metal (átomo 1) y su esfera de coordinación (átomos 2, 5, 6, 9, 10 y 13) son los de menor fluctuación.
El comportamiento de los ligandos a través de los frames nos permite evaluar qué tantas diferencias presentan entre ellos cuando se encuentran en el sistema de simulación. De esta manera, se observa en los boxplots mostrados en la Figura 13, que presentan ligeras diferencias. Prácticamente no hay interacciones intramoleculares (intraHB), el RMSD de Me-DFO se encuentra en torno de 1 Å, el radio de giro se encuentra alrededor de 4.65-4.85 Å, el área de la superficie molecular (MolSA) alrededor de 520 Å2 considerando el radio de van der Waals, el área superficial de una molécula accesible por una molécula de agua (SASA) entre 300 a 320 Å2 y finalmente, el área de la superficie accesible al solvente en una molécula aportada solo por átomos de oxígeno y nitrógeno (PSA), en torno de 200 Å2.
En general, nuevamente encontramos que estos se comportan de manera análoga, a pesar de presentar interacciones intermoleculares diferentes como se observó en la Figura 10.
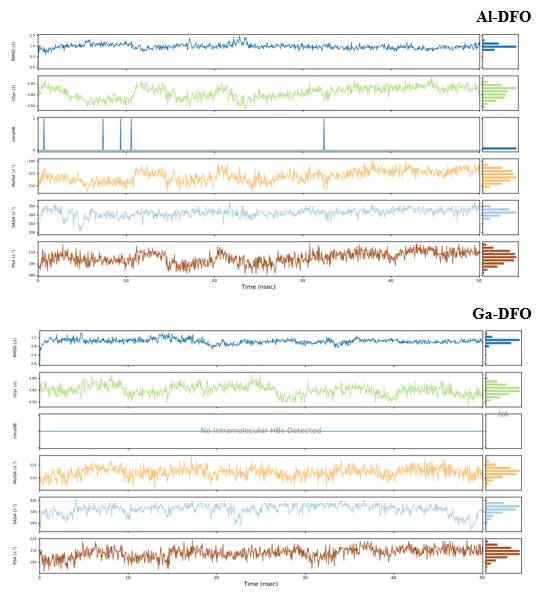
Figura 13.
CONCLUSIONES
Los cálculos realizados con DFT nos han permitido obtener una estructura con RMSD 0.8 Å con respecto del cristal de Fe-DFO, de esta manera, el nivel de cálculo adoptado ha sido suficiente y nos sirvieron en la optimización de las estructuras de Al-DFO y Ga- DFO. Encontramos que los complejos de aluminio y galio presentan diferencias en reactividad química con Fe-DFO, sin embargo, obtenemos energías de afinidad análogas con la proteína FhuE, esto parece indicar que el sideróforo DFO logra apantallar el intercambio de Fe3+ por Al3+ o Ga3+. Descubrimos que durante la simulación de dinámica molecular los complejos Al-DFO y Ga-DFO se comportan de forma análoga con el Fe- DFO, como se puede observar en las fluctuaciones de los átomos del ligando (L-RMSF) o en las interacciones intermoleculares con los residuos de aminoácidos, sin embargo, el tiempo de interacción es diferente y en el caso de Al-DFO, se hace predominante el enlace de hidrógeno, mientas que en Ga-DFO ocurre en menor magnitud, asemejándose más al caso de Fe-DFO. En conclusión, estos resultados nos permiten confirmar que FhuE presenta un reconocimiento molecular de Al-DFO y GaDFO análogo al Fe-DFO, por ello, el diseño de nuevos complejos metal-sideróforo podrían ser una vía exitosa para la aplicación del efecto caballo de Troya en A. baumannii.

