











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
INTRODUCCION
La anemia aplásica (AA) es una enfermedad infrecuente, caracterizada por una insuficiencia de la medula ósea con potencial mortalidad(1), con una incidencia aproximada de 2,7 casos de AA por millón de personas por año(2,3), con mayor presentación entre los 15 a 25 años y entre los 65 a 69 años(4); por su etiología puede ser de causa estructural o adquirida, siendo la más frecuente la adquirida, y de esta un 70 a 80% de causa idiopática(5).
Esta enfermedad presenta un curso clínico variado proporcional a la pancitopenia producida, caracterizada por anemia, leucopenia y plaquetopenia, cuyo diagnóstico se determina con la clínica y el hallazgo de una médula ósea hipocelular en la biopsia, sin evidencia de infiltración neoplásica ni de síndrome mieloproliferativo(5,6).
Su fisiopatología aún resulta desconocida, se plantea diversas hipótesis de las que destaca un cuadro de destrucción de células madre hematopoyéticas por una respuesta inmunitaria activada de manera inapropiada en el contexto de un desencadenante(7). Esta frecuentemente relacionada con factores físicos o químicos como medicamentos, el benceno, radiación ionizante, los insecticidas y también enfermedades autoinmunes e infecciones virales(4,5).
El virus linfotrópico humano tipo 1 (HTLV-1) es un retrovirus frecuentemente asociado a patologías neoplásicas como linfoma - leucemia de células T del adulto y la mielopatía asociada a HTLV-1 o paraparesia espástica tropical(8); sin embargo, no es considerado dentro de los virus relacionado con la fisiopatología de AA(6,9).
Por lo expuesto, el objetivo del presente trabajo fue informar acerca de un caso de anemia aplásica asociada a infección por el virus HTLV-1, se discute el caso y se revisa la literatura.
REPORTE DE CASO
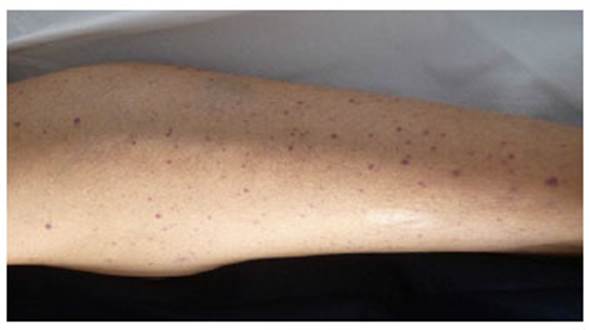
Paciente mujer de 28 años con tiempo de enfermedad de cuatro semanas caracterizado por petequias a predominio de miembros inferiores generalizándose en todo el cuerpo. Dos semanas antes de su ingreso presenta gingivorragia, deposiciones melénicas de moderada cantidad en dos ocasiones, malestar general, sensación de alza térmica a predominio nocturno, diaforesis nocturna e hiporexia. Al examen físico se encontró despierta conectada con el entorno, presentó una presión arterial de 110/40 mmHg, frecuencia cardiaca de 100/minuto y temperatura de 39,5°C. Se encontró lesiones petequiales en la piel distribuida en todo el cuerpo a predominio de miembros inferiores (Figura 1), palidez marcada en piel y mucosa, conjuntivas pálidas, no hubo lesiones equimóticas. No se encontró adenopatías, ni visceromegalia. El resto del examen fue normal.
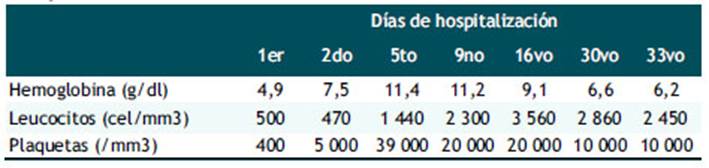
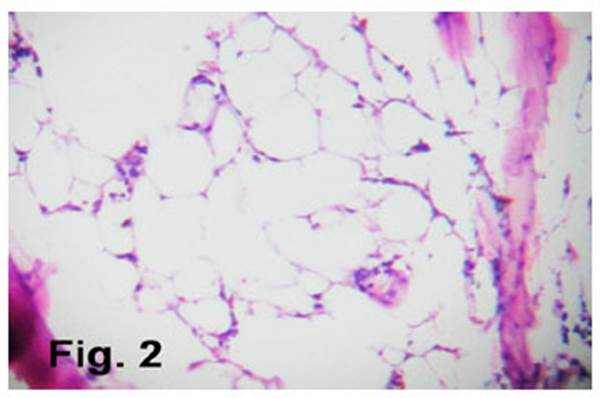
Los exámenes auxiliares al ingreso se encontraron leucocitos 500 cel/mm3, plaquetas 400/mm3, hemoglobina 4,9g/dl, reticulocitos 0,3%, urea 7,3 mg/dl, creatinina 1,18mg/dl, glucosa 108mg/dl, tiempo de protrombina 14,3 e INR 1,24. Ante la evidencia de una pancitopenia severa se le administró dos unidades de paquete globular, seis unidades de plaquetas, gammaglobulina 0,5g/kg/día, dexametasona 4mg c/8h EV (endovenoso) y por el síndrome febril asociado se le agregó ceftazidima 1g c/8h EV, ciprofloxacino 400mg c/12h EV, omeprazol 40 mg c/12h EV y lactulosa 10cc c/8h vía oral. Hospitalizada se le realizó una serie de hemogramas control (Tabla 1) y exámenes auxiliares que resultaron BK (-), sífilis (-), VIH (-), HTLV-1 (+), VHB (-), VHC (-). Para confirmar HTLV-1 se le solicitó por segunda vez prueba de ELISA además de un Western blot que resultaron positivo. Por ese motivo se añadió a la terapia cotrimoxazol (Sulfametoxazol + trimetoprima) 800/160 mg c/24h, fluconazol 150 mg c/7dias y lamivudina 150 mg c/12h. Los resultados de los hemogramas demostraron una evolución desfavorable a pesar del tratamiento recibido por lo que se le inició ciclosporina 50mg c/12h y ácido fólico 5mg/8h vía oral. También realizó un aspirado de medula ósea, el cual dio como resultado una marcada hipocelularidad (Figura 2). Hasta el momento de la comunicación se estaba evaluando la posibilidad de referir a la paciente a un centro de mayor complejidad para un transplante de progenitores hematopoyéticos alogénico pues el tratamiento inmunodepresor no brindaba los resultados deseados.
DISCUSIÓN
Las manifestaciones clínicas de la aplasia medular se relacionan directamente con la citopenia en las tres líneas germinales y dependen de su grado observándose principalmente anemia con reticulocitopenia, leucocitopenia y trombocitopenia manifiesta con petequias, equimosis, gingivorragia y epistaxis que se presentaron en el caso de la paciente(5).
El diagnóstico se plantea por la detección de pancitopenia persistente e inexplicable reducción de celularidad hematopoyética, sin evidencia de infiltración neoplásica, ni de síndrome mieloproliferativo en la muestra patológica de medula ósea; además, clínicamente no debemos de encontrar ganglios linfáticos agrandados, sin esplenomegalia ni hepatomegalia(1,6,10), como en el caso que reportamos.
Se tiene como principal diagnostico diferencial al síndrome mielodisplásico(10), sin embargo, la ausencia de displasia en particular de la megacariopoyesis y la ausencia de células blásticas, así como, la falta de aumento del número de células CD34 y CD117 positivas mediante inmunohistoquímica nos puede apoyar en el diagnóstico de AA(6).
Si bien son múltiples los factores desencadenantes como agentes químicos y físicos, los virus son una causa considerable. Las especies de diferentes familias de virus como parvovirus, herpes virus, virus Ebstein Barr, retrovirus, entro otros virus pueden afectar la función celular de la medula ósea(7). Dentro de los retrovirus se encuentra el HTLV - 1, que es muy frecuente en Perú(11). Es un virus intracelular que ataca a los linfocitos T(5), ocasionando leucemia-linfoma de células T, enfermedad neuroinflamatoria desmielinizante conocida como paraparesia espástica tropical; empero, no hay reportes de su posible relación con la fisiopatología de la AA (6,9), como sospechamos que ocurrió en el presente caso, debido a que el virus tiene capacidad de generar disturbios inmunológicos e infectar células mieloides y linfoides(12).
Se estima que HTLV-1 puede infectar aproximadamente a 10- 20 millones de personas en todo el mundo y su trasmisión se da mediante el contacto con fluidos corporales que contienen células infectadas como el intercambio de agujas entre consumidores de drogas, las transfusiones sanguíneas, las relaciones sexuales, la lactancia materna e incluso la donación de órganos(5,11). La seroprevalencia tiende a aumentar con la edad y las mujeres tienen casi el doble de probabilidades de infectarse que los hombres(5).
La infección por HTLV-1 se diagnostica por medio de pruebas serológicas, siendo la que más se utiliza la prueba de inmunoensayo ligado a enzimas (ELISA)(13); sin embargo, para confirmar el diagnóstico y para distinguir entre HTLV-1 y HTLV-2 se recomienda realizar una prueba de confirmación serológica como Inmunoensayo en línea (INNO-LIA), Western Blot o confirmación molecular (PCR)(8,13). En el caso se realizó el diagnóstico de infección con la prueba de ELISA y Western Blot.
En el contexto de AA, los pacientes son susceptibles a infecciones y complicaciones hemorrágicas lo cual puede llegar a ser fatal si no es tratada a tiempo(14), por lo que es importante determinar la gravedad haciendo uso de los criterios de Camitta(15), catalogándose como severa cuando se cumplen dos de los siguientes: recuento absoluto de neutrófilos <500/μL, recuento de reticulocitos corregido <1%, y recuento de plaquetas <20 000/ μL(4), en nuestro caso se cumplieron los tres criterios.
El tratamiento se enfoca a la restauración de la hematopoyesis normal mediante el trasplante de células madre hematopoyéticas alogénico, terapia inmunosupresora con globulina antitimocito y ciclosporina, y ciclofosfamida en dosis altas sin trasplante de médula ósea(4,5,15). En el caso que reportamos se usó terapia inmunosupresora con ciclosporina que no logró conseguir los resultados esperados por lo que se planteó la referencia de la paciente a un centro de mayor complejidad para trasplante de células madre hematopoyéticas.
La terapia de apoyo ante la sospecha de AA debe de incluir transfusiones teniendo como objetivo evitar el sangrado; además, de tener una cobertura antibiótica amplia ante una posible sepsis causada por bacterias u hongos como Aspergillus sp o Pneumocistys jiroveci que constituyen causas frecuentes de muerte por AA(5), lo cual se realizó en el presente caso haciendo uso de cotrimoxazol y fluconazol.
Se recomienda que ante el diagnostico HTLV-1 prestar atención a la familia y su seguimiento correspondiente pues aproximadamente un tercio de los familiares cercanos de una persona infectada con HTLV-1 también son portadores del virus(11). La prevención para HTLV-1 sugiere evitar la lactancia materna en las zonas endémicas, selección adecuada de donantes de sangre; así como, una vida sexual saludable debido a que no hay un tratamiento específico para esta infección(16).
CONCLUSIONES
Se presentó un caso inusual de anemia aplásica grave que tuvo como posible desencadenante una infección por el HTLV- 1, agente viral frecuente en nuestro medio que no tiene tratamiento específico; sin embargo, el tratamiento de AA se enfoca en la reposición de la función hematopoyética y prevención de complicaciones por la pancitopenia

