











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
El coronavirus de tipo 2, causante del síndrome respiratorio agudo severo (SARS-CoV-2) es el agente causal de la COVID-19, enfermedad infecciosa ampliamente extendida y responsable de la pandemia que inició en el 2020 1,2. Desde un principio existió controversia acerca de la denominación y terminología para referirse a este virus. Así, en febrero del 2020, Jiang et al. 3, propusieron un cambio de nombre para el coronavirus recién surgido en Wuhan, China. Para ellos la denominación SARS-CoV-2, otorgada por el Comité Internacional de Taxonomía Viral (ICTV por sus siglas en inglés), no era la apropiada y sugirieron cambiarla a coronavirus humano 2019 (HCoV-19), argumentando científicamente que el virus se produce de forma natural y es diferente de otros virus similares al SARS-CoV. Además, dada la probabilidad de una atenuación futura hacia una forma menos patógena, predijeron que el uso del nombre SARS-CoV-2 tendría efectos adversos, tanto sociales como económicos 3.
Asimismo, la diversidad genética de SARS-COV-2 amerita un incesante estudio para su comprensión, análisis y seguimiento. La variabilidad del virus ha demostrado ser constante y es probable que su carrera evolutiva nos lleve hacia la variante Omega o aún más allá. Las estrategias para contener nuevas variantes aún están basadas en las medidas de salud pública conocidas: uso de mascarillas, distanciamiento social, preferencia de espacios abiertos o ventilados, más que a cierre de fronteras; que, por ahora, han sido apropiados sólo para los países que han instaurado la estrategia del COVID-cero. De este modo, la variabilidad genética del virus generando nuevas variantes representa un continuo desafío para la humanidad, que tendrá que enfrentar hacia “la nueva normalidad'' 4.
Los efectos sociales y económicos de la COVID-19 han sido inconmensurables alrededor del mundo y la futura atenuación del virus parece ser una utopía aún. Esto debido a la reciente aparición de nuevas variantes de interés derivadas de linajes ampliamente distribuidos, que en un inicio se extendieron probablemente por un efecto fundador, pero ante su extensa diseminación, han surgido de forma convergente en distintas áreas geográficas 2,5,6. Ante ello, los cambios genéticos del virus dieron lugar a múltiples denominaciones o términos como: mutación, variante, linaje y cepa; los cuales se usan frecuentemente y de manera indistinta para describir los cambios que ha sufrido el virus durante la pandemia 7-9.
Para interpretar la gran cantidad de información que se genera diariamente sobre las variaciones genéticas del virus y su impacto en la salud pública mundial, se debe tener clara la terminología empleada para definir la diversidad genómica en SARS-CoV-2. Por este motivo, el presente artículo pretende describir la nomenclatura genómica utilizada para la comunicación general y científica del SARS-CoV-2, asimismo, se describen las mutaciones más importantes, evolución, origen y variantes de interés y preocupación del virus.
METODOLOGÍA
Se realizó una revisión narrativa de literatura procedente desde la identificación del virus hasta diciembre del 2021. La búsqueda y análisis de la información se realizó en un rango personalizado de tiempo, desde 01 de septiembre de 2019 al 15 de diciembre de 2021, utilizando los descriptores Medical Subject Headings (MeSH) enlazados a términos libres: SARS-CoV-2, Evolution, Genome, Viral, Mutation, Sequence Analysis, Phylogeny, SARS-CoV-2 variants, Lineages, Clades, Molecular Epidemiology, COVID-19. A partir de la información obtenida, se realizó una revisión bibliográfica de un total de 1 410 artículos publicados en las bases de datos MEDLINE/ PubMed, SciELO y LILACS, sin restricciones de idioma. También se consideraron documentos oficiales publicados en repositorios de pre impresión, informes de la Organización Mundial de la Salud (OMS), de los Centros para el Control y Prevención de Enfermedades de los Estados Unidos, Virological, PANGO lineages, GISAID y del Departamento de Salud y Asistencia Social del Reino Unido.
La selección de las fuentes filtradas se realizó con la ayuda del gestor de referencias Mendeley Reference Manager. Se excluyeron las fuentes duplicadas, poco relevantes y las que, a la evaluación del título y resumen, no presentaron contenido específico del objetivo de estudio. Después del cual quedaron 74 fuentes seleccionadas para la revisión.
NOMENCLATURA GENÓMICA
La nomenclatura genómica es técnica y con frecuencia desconocida para la población general y parte de la comunidad científica. Se habla de mutación cuando ocurre un cambio en la secuencia de nucleótidos que conforman el genoma del virus. Así, por ejemplo, la mutación D614G en la glicoproteína spike del SARS-CoV-2, sustituye al ácido aspártico en la posición 614 por glicina. Lo mismo sucede con las mutaciones N501Y, E484K, y otras 10,11.
Los genomas que difieren en secuencia a menudo se denominan variantes, que son el producto de muchas mutaciones acumuladas o importantes, de modo que, dos variantes pueden diferir en una o más mutaciones. Las mutaciones en el genoma viral se conservan y acumulan en las posteriores replicaciones del virus, lo que permite realizar comparaciones para evaluar su parecido entre sí y generar, por métodos bioinformáticos, árboles filogenéticos que permiten rastrear su origen 12. Estas variantes se ubican frecuentemente dentro de linajes, un término que se suele utilizar como sinónimo de variante, aunque en otros escenarios taxonómicos un linaje pueda albergar dos o más variantes 13. Las variantes a su vez pueden agruparse en clados diferenciados que adoptan una nomenclatura particular según el sistema de clasificación. Cabe aclarar que el término cepa no presenta una connotación genómica, más bien microbiológica, referida al aislamiento viral a partir de una muestra, la misma que ha sido caracterizada 14,15 (Figura 1).
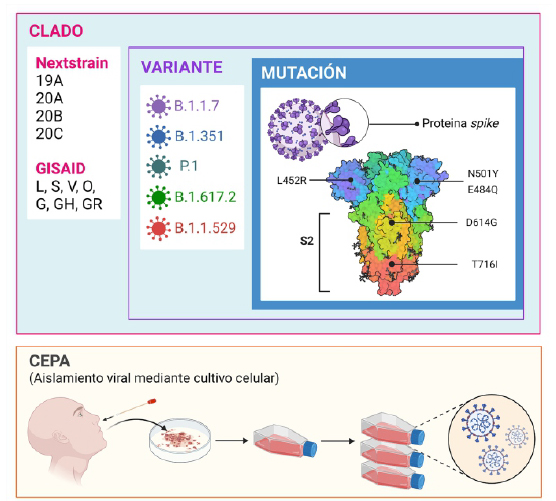
Figura 1 Terminología genómica utilizada con frecuencia para referirse a los distintos niveles de organización del SARS-CoV-2
A la fecha existen más de 300 linajes descritos para SARS-CoV-2 16. La nomenclatura de estas es muy heterogénea en la actualidad y pueden generar confusión. La Public Health England (PHE) considera aspectos epidemiológicos, inmunológicos y propiedades patógenas para clasificar las variantes en Reino Unido. La PHE utiliza el término “Variante Bajo Investigación”, asociado al año, mes y número. Si el comité de expertos considera necesario, una variante bajo investigación puede cambiar por “variante de preocupación” (VDP). Esto sucedió con la variante conocida como linaje B.1.1.7 o variante del Reino Unido; y que posteriormente fue denominada variante de preocupación el 18 de diciembre de 2020, debido a su rápida expansión en ese país 17.
Actualmente, la plataforma Nextstrain define veintidós clados relacionados filogenéticamente para clasificar a las variantes del SARS-CoV-2, nombrados en función de los tres últimos números del año estimado en el que emergieron (19, 20 y 21) y seguido de una letra genérica (A, B, C). Así, el primer clado se denomina 19A y es considerado el clado raíz del que surgen los demás (figura 1). Apareció en Wuhan y junto al 19B dominaron el brote temprano. El clado 19A alcanzó una frecuencia global de 47 a 65 % en enero de 2020. El clado 20A surgió a partir del 19A, dominando el brote europeo en marzo del 2020 y desde entonces se ha extendido por todo el mundo. Los clados 20B y 20C son grandes subclados genéticamente distintos del 20A, también distribuidos globalmente. Los clados 20D al 20I aparecieron durante el verano del 2020 e incluyen dos VDP con mutaciones distintivas en la proteína S: N501Y, 20I (Alfa, V1) y 20H (Beta, V2) y a finales de diciembre del 2020, emergió el clado 20J que incluye a la VDP Gamma o linaje P.1. Los clados 21A a 21K incluyen dos VDP (Delta y Ómicron) y varias VDI (Figura 2) 7,18.
Por su parte, la base de datos de GISAID, iniciativa creada en un principio para depositar genomas del virus Influenza, para la nomenclatura del SARS-CoV-2 y su clasificación en clados, utiliza letras reales en base a mutaciones de marcadores, en lugar de letras genéricas (A, B) como ocurre en Nextstrain. Por ejemplo, S-D614G es uno de varios marcadores genéticos que caracterizan a un nuevo clado que aumentó bruscamente desde febrero de 2020 y la letra G fue elegida para nombrarlo en ese momento. Actualmente GISAID clasifica las secuencias del SARS-CoV-2 en ocho clados que son: S, L, O, V, G, GH, GR y GV (figura 2), donde los cuatro últimos albergan la mutación D614G en la glicoproteína spike del virus que está presente en las variantes de preocupación recién reportadas 8.
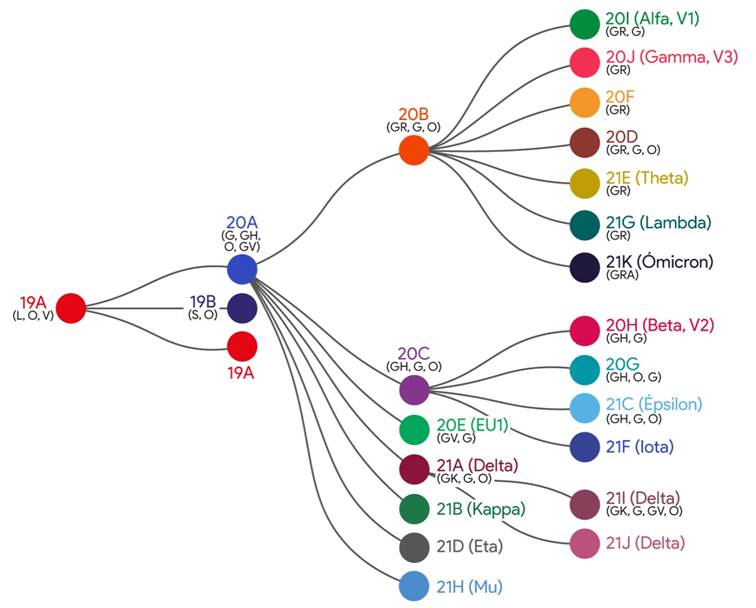
Figura 2 Relación filogenética de clados del SARS-CoV-2, según las definiciones de Nextstrain (clados en color) y GISAID (clados entre paréntesis y en orden descendente de frecuencias observadas). Datos hasta diciembre del 2021. Adaptada de Nextclade. Nextstrain. 2021. Disponible en: https://clades.nextstrain.org/. Referencia 7
Existe un sistema jerarquizado y dinámico para definir la aparición de nuevos brotes locales con importancia epidemiológica, utilizando la herramienta de línea de comandos y aplicativo web Phylogenetic Assignment Of Named Global Outbreak Lineages (Pangolin) 19. Esta herramienta se basa en la nomenclatura propuesta por Rambaut et al 14, que asignan linajes a las secuencias de consulta teniendo en cuenta múltiples fuentes de información filogenética, epidemiológica, así como una variedad de metadatos asociados a la secuencia. Pangolin indica relaciones evolutivas entre los linajes del SARS-CoV-2, en el que cada carácter sucesivo denota un subgrupo del anterior estipulado por letras, ejemplo, linaje B.1.1.7. Creemos que este sistema es el más apropiado, pues elimina los nombres que asocian a una variante o linaje con el país o la región en la que se identificó, evitando designarlas como variante británica, sudafricana, brasileña entre otras 19.
La definición de cepa es el término que ha dado lugar a mayor confusión por algunos medios de divulgación 12,20. En este sentido, la OMS se refiere a la variante del Reino Unido de la siguiente manera: “más del 50 % de los aislamientos se identificaron como cepa variante” 21. En otro informe indica que “los estudios en células respiratorias humanas y en modelos animales demostraron que, en comparación con la cepa del virus inicial, la cepa con la sustitución D614G tiene una mayor infectividad y transmisión” 1. Cómo se lee en ambos informes, la OMS identifica claramente el término cepa, para referirse al producto de un aislamiento viral en cultivo microbiológico, asegurando su pureza y ausencia de subpoblaciones o “cuasi especies” que suelen originarse una vez ingresan al huésped susceptible. Esta definición se complementa con lo dicho por Fauquet et al. 22, quienes sostienen que “las cepas son virus que pertenecen a la misma especie y difieren en características biológicas, serológicas y moleculares estables y hereditarias”.
MUTACIONES DEL SARS-CoV-2
En SARS-CoV-2, como en otros microorganismos, las mutaciones generalmente surgen de forma independiente y reciben el nombre de homoplasias, término referido al cambio evolutivo paralelo que hace que los organismos presenten un mismo carácter adquirido de manera independiente. De estas homoplasias se han identificado 198 mutaciones recurrentes en el genoma del SARS-CoV-223.
La mutación más frecuente en los genomas secuenciados en todo el mundo es una transversión que afecta a la adenosina de nucleótido 23403, y la transforma en una guanosina (A23403G). Esta mutación define el llamado clado G de los genomas del SARS-CoV-2, prevalente en Europa, Oceanía, América del Sur y África. El efecto de esta mutación es un cambio de aminoácidos que produce el ya mencionado D614G. El análisis de datación molecular estimó la aparición de este clado entre mediados y finales de enero del año 2020 24. Asimismo, tres mutaciones denominadas C14408T, C241T y C3037T muestran una frecuencia similar con A23403G (tabla 1). De modo que estas cuatro mutaciones casi siempre coexisten en los mismos genomas, definiendo el clado G, principal clado observado en la población viral 25.
Una de las primeras mutaciones de interés que adquirió SARS-CoV-2 fue la sustitución de D614G en la proteína spike. Esta mutación proporciona al virus la ventaja de mayor infectividad y transmisión en modelos animales, cultivos celulares y humanos 26. Un estudio en hámsters infectados con D614 y G614, encontró títulos infecciosos más altos del SARS-CoV-2 con la mutación G614 en muestras de lavados nasales y de tráquea, pero no en los lavados pulmonares. Estos hallazgos respaldan la evidencia clínica de que la mutación G614 aumenta la transmisión al tener cargas virales más altas en el tracto respiratorio superior de pacientes con COVID-19 26,27. Sin embargo, es poco probable que reduzca la capacidad de neutralización de las vacunas en curso 28, ya que, cuando se enfrentaron los sueros de los hámsters infectados con (D614), estos neutralizaron modestamente al virus mutado (G614) 10. Resultados similares se encontraron con sueros de ratones inmunizados con la proteína spike enfrentados a un pseudovirus G614 28, y en cultivos celulares humanos, donde a pesar de que G614 exhibe mayor capacidad de infección y replicación en las células epiteliales de las vías respiratorias, mantiene una morfología y propiedades de neutralización in vitro similares al virus ancestral de tipo salvaje 27.
Tabla 1 Mutaciones más frecuentes del SARS-CoV-2 durante el primer semestre de la pandemia.
| Coordenadas genómicas | Efecto en la proteína / UTR | Número de muestras | Tipo | Región genómica |
|---|---|---|---|---|
| A23403G | S:D614G | 36 500 | Cambio de aa por SNP | Proteina Spike |
| C14408T | NSP12b:P314L | 36 444 | Cambio de aa por SNP | Proteína no estructural 12, mutación por desplazamiento de marco de lectura post ribosomal (ARN pol dependiente de ARN) |
| C3037T | NSP3:F106F | 36 384 | SNP silente | Proteína no estructural 3 (fosfoestereasa predicha) |
| C241T | 5´UTR:241 | 36 007 | SNP en 5´UTR | 5’ región no traducida |
| GGG28881AAC | N:RG203KR | 14 095 | Cambio de aa por SNP triplete | Proteína nucleocápside |
| G25563T | ORF3a:Q57H | 10 929 | Cambio de aa por SNP | Proteina ORF3a |
| C1059T | NSP2:T85I | 8 451 | Cambio de aa por SNP | Proteína no estructural 2 |
| G11083T | NSP6:L37F | 5 507 | Cambio de aa por SNP | Proteína no estructural 6 (proteína transmembrana) |
| C14805T | NSP12b:Y446Y | 4 505 | SNP silente | Proteína no estructural 12, mutación por desplazamiento de marco de lectura post ribosomal (ARN pol dependiente de ARN) |
| T28144C | ORF8:L84S | 3 804 | Cambio de aa por SNP | Proteína ORF8 |
| G26144T | ORF3a:G251V | 3 792 | Cambio de aa por SNP | Proteina ORF3a |
| C8782T | NSP4:S76S | 3 743 | SNP silente | Proteína no estructural 4 |
| A20268G | NSP15:L216L | 2 479 | SNP silente | Proteína no estructural 15 (endo ARNasa) |
| C18060T | NSP14:L7L | 1 813 | SNP silente | Proteína no estructural 14 (3´- 5´ exonucleasa) |
| C23731T | S:T723T | 1 799 | SNP silente | Proteina Spike |
| G10097A | NSP5:G15S | 1 798 | Cambio de aa por SNP | Proteína no estructural 5 (proteasa) |
| A17858G | NSP13:Y541C | 1 780 | Cambio de aa por SNP | Proteína no estructural 13 |
| C17747T | NSP13:P504L | 1 736 | Cambio de aa por SNP | Proteína no estructural 13 |
| C2558T | NSP2:P585S | 1 701 | Cambio de aa por SNP | Proteína no estructural 2 |
| A2480G | NSP2:I599V | 1 615 | Cambio de aa por SNP | Proteína no estructural 2 |
| aa: aminoácido; SNP: polimorfismo de nucleótido único; UTR: región no traducida |
Modificado de Front Microbiol. 2020;11. Referencia 25.
Algunas mutaciones afectan al motivo de unión al receptor de la proteína spike del SARS-CoV-2 (RBM), el cual es una región altamente variable de S. En esta se ha reportado una mutación denominada N439K. Se ha demostrado que la proteína N439K tiene una mayor afinidad de unión al receptor ACE2 humano, y los virus que albergan N439K tienen similares propiedades in vitro, capacidad de replicación y capacidad de causar infecciones con resultados clínicos similares en comparación con el tipo salvaje. Así mismo, la mutación N439K confiere resistencia contra varios anticuerpos monoclonales neutralizantes, incluyendo uno autorizado para uso de emergencia por la FDA y reduce la actividad de algunos sueros policlonales de personas recuperadas de la infección (Figura 3) 29.
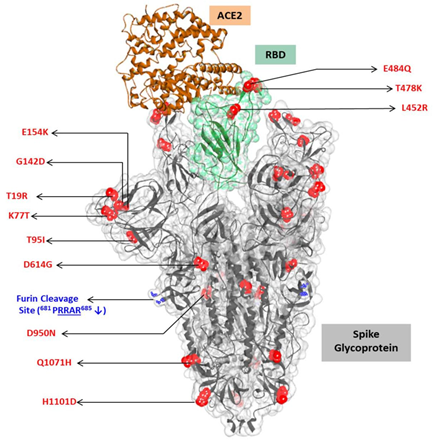
Figura 3 Mutaciones más frecuentes en la proteína spike del SARS-CoV-2. Figura tomada de Microorganisms. 2021;9(7):1542. Referencia 11
La aparición de una nueva variante denominada CAL.20C que se define por cinco mutaciones concurrentes: ORF1a: I4205V, ORF1b: D1183Y, S: S13I; W152C; L452R. Una característica que comparte con las cepas del Reino Unido y Sudáfrica, ambas de gran interés clínico y científico. Aunque esta variante fue detectada en julio de 2020 y después observada en octubre de 2020, en diciembre de 2020 ya representaba cerca del 36% de muestras de Los Ángeles. El incremento de esta variante coincide con el pico en casos en California meridional 30.
Las mutaciones de evasión inmunológica que mantienen la virulencia y la aptitud, como N439K, y las variantes de rápida diseminación cómo CAL.20C pueden surgir en SARS-CoV-2, en cualquier parte del mundo, sobre todo en aquellas zonas donde existe un elevado número de contagios. Esto destaca la necesidad de una vigilancia molecular continua para guiar el desarrollo y el uso de vacunas y terapias 30.
EVOLUCIÓN Y ORÍGEN DE SARS-CoV-2
Los primeros análisis genómicos del SARS-CoV-2 revelaron que este comparte más del 96 % de su genoma con un coronavirus que afecta a murciélagos 31. Boni et al. 32, tras intentar reconstruir el árbol genealógico del virus, proponen un único linaje ancestral para el SARS-CoV-2 y RaTG13, y que este linaje en común se separó hace 40 y 70 años aproximadamente, lo que significa que el SARS-CoV-2 lleva circulando muchas décadas de manera silente y no detectada entre la población de murciélagos 32. Estos hallazgos sugieren que por el largo periodo de divergencia, existen muchos linajes virales en murciélagos con potencial zoonótico y que aún no han sido muestreados (Figura 4).
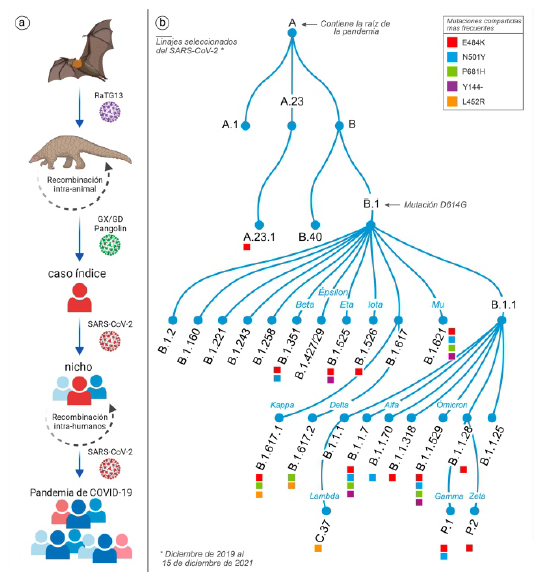
Figura 4 A) Probable origen del SARS-CoV-2. B) Evolución del SARS-CoV-2 en linajes según la nomenclatura Pango y mutaciones compartidas más frecuentes. Adaptado de las referencias 71-73.
Al comienzo de la pandemia de la COVID-19, los genomas del SARS-CoV-2 más frecuentes recuperados y notificados al GISAID pertenecen al clado L (figura 1). El clado S, apareció a principios de 2020, incluyendo los primeros genomas del virus mutado. Posterior a ello, aparecían los clados O y V (mutado en NSP6 y ORF3a) a mediados de enero de 2020, aproximadamente al mismo tiempo que el clado G original. La expansión mundial del clado G alcanzó el 50 % en marzo de 2020, dividiéndose en los subclados GH, GR a finales de febrero y luego en GV 8. Los esfuerzos de secuenciación localizados principalmente en América del Norte y Europa, han demostrado una frecuencia creciente de genomas G, GH y GR, convirtiéndose gradualmente en las secuencias más representadas en GISAID 9.
Actualmente, los genomas más prevalentes en América del Norte pertenecen al clado GH, que representan más del 50 % de las secuencias enviadas a GISAID. En Europa y América del Sur, el clado más predominante es GR, mientras que en Oceanía parece haber la coexistencia más equilibrada de todos los clados observados hasta la fecha 33. África muestra una prevalencia del clado G, y Asia caracterizada inicialmente por sus secuencias de referencia (clado L y S), con el transcurso de la pandemia fueron reemplazadas por genomas pertenecientes a los clados G, GH y GR, ganando terreno en el continente asiático desde principios de marzo de 2020 25,33.
A finales de 2020, se ha podido apreciar el incremento de la diversidad genómica del virus. Por ejemplo, países europeos tienen una prevalencia de genomas de GH (Dinamarca, Francia), mientras que otros muestran cifras más altas del clado GR (Reino Unido, Portugal). El clado que predomina actualmente en los Estados Unidos de América es GH, de igual manera en Israel y Arabia Saudita, mientras que en Rusia y Brasil los genomas más comunes pertenecen al clado GR. Se aprecia un aumento con el tiempo en los genomas del clado G, y sus derivados GH y GR, emparejados por una desaparición gradual de los clados L y V (figura 1). El clado S, aunque declina, parece seguir representando una minoría significativa de genomas secuenciados, especialmente en los Estados Unidos de América y España 25.
¿Existe evolución por selección natural en SARS-CoV-2?
Para evaluar una nueva variante de SARS-CoV-2, es importante conocer si alcanzó su predominancia a través de la selección natural o por eventos fortuitos como un efecto fundador tal como pudo ocurrir con SARS-CoV34. Si la evidencia sugiere selección natural, se debe evaluar el tipo de mutación, el beneficio adaptativo y su probable influencia sobre la transmisibilidad, antigenicidad o virulencia.
Los virus de ARN como los rotavirus, coronavirus, retrovirus y orthomyxovirus, tienden a producir mutaciones rápidamente a medida que copian su material genético dentro de la célula huésped. Esto debido a que las enzimas que copian el ARN viral son propensas a cometer errores y tienen una menor capacidad de reparación comparadas con las polimerasas de ADN. Estas mutaciones por lo general son deletéreas, y si la diseminación de los virus es limitada y controlada, reducirá su variabilidad genética, lo que en ocasiones puede conducir a un fenómeno llamado cuello de botella, donde una mutación desfavorable puede extinguir al virus, tal como se sospecha sucedió con SARS-CoV, que después de que comenzará a circular en los humanos, una deleción de 29 nucleótidos en su genoma, podría haber frenado su propagación 23,35.
Se estima que la tasa de mutación del SARS-CoV-2 es aproximadamente uno a dos nucleótidos por mes 36. Desde marzo de 2020, la mutación D614G en la proteína spike se notificó de forma independiente y simultánea en múltiples regiones del mundo 26. En España las primeras variantes se ramificaron en el clado 19B (virus D614), que fue el clado más prevalente durante las primeras semanas de marzo, lo que apunta a un efecto fundador. Sin embargo, desde mediados de marzo hasta junio de 2020, los virus portadores de G614 (clados 20A, 20B y 20C) superaron las variantes D614 en España, probablemente como consecuencia de una ventaja evolutiva 37. Aparentemente esto indica una evolución convergente y selección natural por beneficio adaptativo de G614 al huésped, sin embargo, los esfuerzos de secuenciación posteriores identificaron la mutación G614 en virus en varias provincias chinas a finales de enero. Esto planteó la hipótesis de que la dispersión global de esta mutación podría haber resultado de eventos fundadores casuales, en los que los virus que albergan G614 iniciaron la mayoría de los eventos de transmisión temprana en múltiples ubicaciones 26.
Por otra parte, la mutación N501Y del linaje B.1.1.7 que ha permitido que el SARS-CoV-2 se disemine ampliamente por el Reino Unido, podría deberse a la evolución del virus por selección natural desde un individuo infectado crónicamente. Esto en base a estudios de pacientes inmunodeficientes o inmunosuprimidos infectados crónicamente con SARS-CoV-2, donde se ha informado altas tasas de acumulación de mutaciones durante períodos de tiempo corto 38.
Otra hipótesis sugiere que la terapia con anticuerpos (plasma de convalecencia) en pacientes inmunosuprimidos, induce selección 39. Por lo general, esta terapia se usa cuando la carga viral del paciente es alta o infección crónica. Si la población de virus es inusualmente grande y genéticamente diversa en el momento en que se administra el tratamiento, la presión selectiva creará las circunstancias adecuadas para la rápida fijación de mutaciones en el genoma del virus a través de selección natural intra huésped, un fenómeno ya descrito en la generación de cuasiespecies. Debido a que esta práctica es difundida en algunos países, se espera que la dinámica evolutiva y las presiones selectivas sobre la población de virus intrapaciente sean muy diferentes a las experimentadas en la infección típica 40.
VARIANTES DEL SARS-CoV-2
El grupo interagencial de Estados Unidos de Norteamérica, conformado por el Centro de Control de Enfermedades (CDC, por sus siglas en inglés de: Central Disease Control); el instituto Nacional de Salud (NIH, por sus siglas en inglés de: National Institutes of Health), y la Administración de Alimentos y Medicamentos (FDA, por sus siglas en inglés de: Food and Drug Administration); clasificaron en cuatro tipos las variantes del SARS-CoV-2 con el objetivo de monitorear su impacto potencial en la salud pública: a) variantes que se están monitoreando, b) variantes de interés, c) variantes de preocupación y d) variantes de alta consecuencia 5. Asimismo, la OMS también ha propuesto la clasificación de: variantes de interés y preocupación, aunque pueden diferir con la de USA, asimismo, ha propuesto etiquetas que constan del alfabeto griego para simplificar los debates públicos sobre las variantes: Alfa, Beta, Gamma, etc 6.
Variantes de preocupación (VDP)
Estos virus, además de presentar los atributos de las VDI, es decir: tienen un genoma que contiene mutaciones de implicancias fenotípicas respecto al aislado de referencia, es causa de transmisión comunitaria o de múltiples grupos y ha sido detectada en múltiples países (tabla 2); presentan los siguientes atributos 5:
Variante Alfa (Linaje B.1.1.7)
También llamada variante británica. Es un grupo filogenético que se ha extendido rápidamente desde el sureste del Reino Unido a diferentes países del mundo (tabla 2). Contiene 23 mutaciones, de las cuales 17 SNP definen el linaje 40. El análisis filogenético indica que esta variante había acumulado una gran cantidad de mutaciones antes de su detección a principios de septiembre (tabla 2), lo que sugiere una cantidad significativa de evolución previa, posiblemente en un huésped con infección crónica 38. Tres de las ocho mutaciones identificadas en la proteína spike, tienen efectos biológicos potenciales: N501Y en el dominio de unión al receptor (RBD), P681H en el sitio de escisión de la furina y la deleción 69/70 en el dominio N terminal de spike, que en conjunto pueden aumentar la infectividad por una mejor afinidad de unión al receptor ACE2 humano, la transmisibilidad y la evasión de la respuesta inmune 40,41. Sin embargo, un buen grupo de datos emergentes indican que tanto los sueros de individuos convalecientes y vacunados con vacunas ARNm no tienen problemas para neutralizar un virus portador de N501Y 42-45.
Tabla 2 Características de las variantes de preocupación (VDP)
| Denominación OMS | Alfa | Beta | Gamma | Delta | Ómicron |
| Linaje Pango | B.1.1.7 | B.1.351 | P.1 | B.1.617.2 | B.1.1.529 |
| Clado GISAID/Nextstrain | GH/20H (501Y.V1) | GH/20H (501Y.V2) | GR/20J (501Y.V3) | G/21A | GRA/21K,21L |
| Primera detección: ubicación/fecha | Reino Unido / septiembre 2020 | Sudáfrica / octubre 2020 | Brasil / noviembre 2020 | India/octubre 2020 | Varios países / noviembre 2021 |
| Fecha de designación | Diciembre 2020 | Diciembre 2020 | Enero 2021 | VDI abril 2021 VDP mayo 2021 | Noviembre 2021 |
| Mutaciones en la proteína spike | 8 | 9 | 10 | 10 | 37 |
| Mutaciones con efectos biológicos potenciales | N501Y, D614G, P681H, deleción 69/70 | K417N, E484K, N501Y, D614G | K417N, E484K, N501Y, D614G | L452R, T478K, D614G y P681R | R203K y G204R |
| Países que han notificado* | 175 | 117 | 88 | 176 | 67 |
VDI: variante de interés, VDP: variante de preocupación. *Datos hasta el 15 de diciembre del 2021. Fuente: Tabla adaptada de la Organización Mundial de la Salud y Topol E. The SARS-CoV-2 variants. Twitter. 2021. Referencias 1,74
Al 28 de diciembre de 2020, esta variante representó aproximadamente el 28 % de los casos de infección por SARS-CoV-2 en Inglaterra, y los modelos genéticos poblacionales sugieren su propagación en un 56 % más rápidamente que otros linajes 41,46. A diferencia de la mutación D614G, que podría haberse beneficiado de los eventos fortuitos de cuello de botella, el linaje B.1.1.7 se expandió cuando los casos de SARS-CoV-2 se generalizaron y aparentemente ha logrado el dominio al superar a una población existente de variantes circulantes. Esto sugiere fuertemente la selección natural de un virus que es más transmisible a nivel de población 40.
Variante Beta (Linaje B.1.351)
Apareció por primera vez en Nelson Mandela Bay, Sudáfrica, en muestras de pacientes que datan de inicios de octubre y en Zambia a finales de diciembre de 2020, momento en el que parecía ser la variante predominante en el país. La variante sudafricana contiene 21 sustituciones de nucleótidos, de las cuales nueve SNP definen el linaje (tabla 2) 47. Entre las mutaciones en la proteína spike se incluye la ya conocida N501Y y dos más preocupantes para la actividad neutralizante de los anticuerpos (E484K, K417N) 48-50. La variante sudafricana a diferencia de la británica, no contiene la deleción 69/70 (tabla 2).
El ocho de enero de 2021, BioNTech y Pfizer, fabricantes de la principal vacuna ARNm (BNT162b2) administrada en el mundo, aseguraron que BNT162b2 es eficaz contra la mutación N501Y, pero sus análisis no incluyeron a las variantes que albergan E484K, por lo que persisten las dudas sobre este punto 43. Por otro lado, un informe publicado en Brasil, dio cuenta de una reinfección por COVID-19 con una variante portadora de la mutación E484K, donde la segunda infección fue más grave que la primera, lo que podría indicar que esta mutación, provocó una respuesta inmune menos eficaz en el paciente 51.
Actualmente, no hay evidencia suficiente que sugiera que esta variante tenga algún impacto en la gravedad de la enfermedad; sin embargo, se sabe por estudios in vitro, que la mutación E484K puede afectar la capacidad de neutralización de algunos anticuerpos monoclonales, policlonales y de los test de diagnóstico molecular 2,52.
Variante Gamma (Linaje P.1)
Descrita por primera vez en Manaus, Brasil, en diciembre de 2020. Este nuevo linaje denominado P.1, que es un alias del B.1.1.28.1 tal como lo describe Rambaut et al 14. Fue identificado por primera vez en cuatro turistas brasileños muestreados durante la detección de rutina en el aeropuerto de Haneda en las afueras de Tokio, Japón (tabla 2) 2. Lo más intrigante del linaje P.1 es su rápida propagación en Manaus, desde 52,2 % (n = 35/67) en diciembre de 2020 a 85,4 % (n = 41/48) en enero de 202153, estas cifras indican un incremento rápido de la frecuencia de casos con esta nueva variante, pudiendo estar asociada a mayor transmisibilidad y peligro de reinfecciones. La mutación K417N al parecer tiene un efecto muy pronunciado en la disminución de la eficacia de anticuerpos y en combinación con la mutación N501Y anularía completamente el efecto de los mismos 54.
Aunque la variante Gamma tiene más mutaciones en la proteína Spike en comparación con la variante Beta, sorprendentemente es significativamente menos resistente a la neutralización por anticuerpos neutralizantes, ya sea adquirida naturalmente o inducida por vacunaciones 55. Además, la neutralización de la Gamma por las vacunas Pfizer BNT162b2 y Oxford-AstraZeneca AZD1222 mostró una reducción de 2,6 veces y 2,9 veces, respectivamente, en relación con la cepa Victoria; considerablemente mejor que la neutralización de la variante Beta, donde se observaron reducciones de 7,6 y 9,0 veces, respectivamente 56.
Variante Delta (Linaje B.1.617.2)
Identificado en octubre de 2020 en el estado de Maharashtra, parte occidental de la India, cuando se realizaba un análisis en muestras recolectadas en esta región 11,57. El linaje incluye tres subtipos principales (B1.617.1, B.1.617.2 y B.1.617.3) y posee mutaciones distintivas comunes: D111D, G142D, L452R, E484Q, D614G y P681R, en la proteína spike, incluido el dominio de unión al receptor (RBD) 11. Dentro de ellos el sublinaje B.1.617.2 (variante delta), responsable de la segunda ola mortal de infecciones por COVID-19 en abril de 2021 en India 58. Se transmite continuamente a una tasa más alta dentro del país y en todo el mundo, desplazando a la variante Alfa, inicialmente establecida 59.
A pesar que inicialmente se consideró como una VDI, su rápida extensión llevó a la OMS a clasificarla como una VDP en mayo de 2021 6. Esta variante contiene diez mutaciones (T19R, (G142D*), 156del, 157del, R158G, L452R, T478K, D614G, P681R, D950N) en la proteína spike, cuatro de ellas son de especial preocupación: L452R, T478K, D614G y P681R; lo que puede asociarse con una alta tasa de transmisión viral y aumento en el escape de neutralización 5,55.
Asimismo, se ha observado que la variante Delta es resistente a la neutralización por algunos anticuerpos monoclonales anti-NTD y anti-RBD, incluido el bamlanivimab, y estos anticuerpos mostraron una unión deficiente a la proteína spike60. Sin embargo, otros estudios recientes sugieren que la mayoría de los individuos que recibieron una vacuna completa de BNT162b2 en dos dosis estarían protegidos contra la variante y la enfermedad asociada 61. Un estudio en Inglaterra informó que la eficacia de dos dosis de la vacuna Pfizer / BioNTech se redujo modestamente del 93,4% contra la Alfa al 87,9% contra la Delta. Pero, la vacuna AstraZeneca, mostró una reducción significativamente mayor en la efectividad contra la variante, 66,1% frente a la Alfa y 59,8% a la Delta 62.
Variante Ómicron (Linaje B.1.1.529)
El primer caso fue reportado en noviembre del 2021, en el país de Botsuana 63. La variante Ómicron presenta 37 mutaciones en la proteína spike, de las cuales 26 son únicas de variante y el resto son compartidas con las variantes Alfa, Beta y Gamma (69-70del, T95I, G142D/143-145del, K417N, T478K, N501Y, N655Y, N679K y P681H); las mismas que le permiten mayor transmisibilidad afinidad de unión viral y escape inmunológico 64,65.
Un estudio previo probó la sensibilidad de 28 muestras de suero de pacientes convalecientes con COVID-19 infectados con la cepa original del SARS-CoV-2 a Ómicron y otras VDP y VDI; observando que la neutralización media ED50 de estos sueros contra Ómicron disminuyó a 66, que es aproximadamente 8,4 veces en comparación con la cepa de referencia D614G (ED50 = 556), mientras que la actividad de neutralización de los otros virus disminuyó sólo alrededor de 1,2-4,5 veces 66.
Variantes de interés (VDI)
Según la OMS, estos virus tienen un genoma que contiene mutaciones de implicancias fenotípicas respecto al aislado de referencia, es causa de transmisión comunitaria o de múltiples grupos y ha sido detectada en múltiples países (tabla 3). Presenta los siguientes atributos 5:
Probable afectación de la transmisión, diagnóstico, terapéutica y respuesta inmunológica.
Evidencia de una mayor proporción de casos o brotes únicos.
Prevalencia o expansión geográfica limitada.
Las VDI merecen un seguimiento especial debido a que presentan cambios genéticos que pueden afectar las características del virus, tales como transmisibilidad, gravedad de la enfermedad, escape inmunológico, escape diagnóstico o terapéutico 6.
Las variantes Eta (B.1.525) e Iota (B.1.526)
Albergan mutaciones de picos clave (B.1.525: A67V, Δ69 / 70, Δ144, E484K, D614G, Q677H, F888L; B.1.526: (L5F *), T95I, D253G, (S477N *), (E484K *), D614G, (A701V *)) y se detectaron por primera vez en Nueva York en noviembre de 2020 y los CDC y la OMS los clasificaron como una variante de interés debido a su posible reducción de la neutralización por anticuerpos, tratamientos y sueros de vacunas 58.
Variante Lambda (C.37)
Denominada “lambda” en su nomenclatura simplificada. Se informó por primera vez en Perú en agosto de 2020 y luego se extendió a la mayoría de los países de América del Sur 67. Se han notificado casos importados en la mayoría de los países europeos desde diciembre de 2020. La aparición de mutaciones asociadas a la pérdida de neutralización en diferentes cepas (L452Q y F490S) hace que la C.37 sea de especial interés. Se ha demostrado que "lambda" tiene una sensibilidad moderadamente reducida a la neutralización por sueros de convalecientes y el anticuerpo monoclonal REGN10987, y una sensibilidad ligeramente reducida a los anticuerpos activados por BNT162b2, mRNA-1273, y CoronaVac, al tiempo que conserva la sensibilidad total hasta REGN10933 68.
Variante Mu B.1.621
Recientemente designado por la OMS en agosto de 2021, se informó por primera vez en Colombia, enero 2021. Presenta varias sustituciones que afectan a la proteína spike, incluyendo los cambios de aminoácidos en la proteína spike (Y144T, Y145S, R346K, E484K, N501Y y P681H) 69.
Las sustituciones en la proteína de pico son comunes, sin embargo, algunas sustituciones distintivas han sido características relevantes, en cuanto a una menor actividad neutralizante del plasma convaleciente y anticuerpos 70. Sin embargo, las implicaciones en términos de infección, transmisión y patogénesis aún se desconocen, por lo que se requieren más reportes que permitan evaluar las funciones biológicas y epidemiológicas 69.
Tabla 3 Variantes de interés
| Denominación OMS | Linaje Pango | Clado GISAID/Nextstrain | Identificado por primera vez | Fecha de designación |
|---|---|---|---|---|
| Épsilon | B.1.427/B.1.429 | GH/21C | USA, marzo 2020 | mar-21 |
| Zeta | P.2 | GR/20B | Brasil, abril 2020 | mar-21 |
| Eta | B.1.525 | G/21D | Varios, diciembre 2020 | mar-21 |
| Theta | P.3 | GR/21E | Filipinas, enero 2021 | mar-21 |
| Iota | B.1.526 | GH/21F | USA, noviembre 2021 | mar-21 |
| Kappa | B.1.617.1 | G/21B | India, octubre 2020 | abr-21 |
| Lambda | C.37 | GR/21G | Perú, diciembre 2020 | jun-21 |
| Mu | B.1.621 | GH/21H | Colombia, enero 2021 | set- 21 |
Adaptado de Tracking SARS-CoV-2 variants. World Health Organization. 2021. Disponible en: https://www.who.int/es/activities/tracking-SARS-CoV-2-variants. Referencia 6.
CONCLUSIONES
La alta diversidad genómica del SARS-CoV-2 se debe cambios en la secuencia de nucleótidos del ARN viral (mutaciones), que pueden conducir a ventajas evolutivas debido a modificaciones estructurales en sus proteínas funcionales como la spike. Cuando cierta acumulación de mutaciones genera cambios en la epidemiología de la enfermedad, da lugar a variantes o linajes, y estos su vez en clados.
Las variantes de interés presentan probable afectación de la transmisión, diagnóstico, terapéutica y respuesta inmunológica; destacan Lambda y Mu, identificadas por primera vez en Perú y Colombia, respectivamente. Las variantes de preocupación presentan evidencia de mayor transmisibilidad, gravedad de la enfermedad e interferencia diagnóstica, susceptibilidad reducida a terapias y disminución significativa de anticuerpos neutralizantes; denominadas Alfa (británica), Beta (sudafricana), Gamma (brasilera), Delta (india) y Ómicron.
La mejor comprensión de la diversidad genómica en SARS-CoV-2 ayudará a tomar medidas de contención adecuadas frente al virus. Se requiere considerar que esta diversidad se puede originar en cualquier parte del mundo, sobre todo en aquellas zonas donde existe un elevado número de contagios. La vigilancia genómica continua y suficiente puede guiar el desarrollo y el uso de vacunas, terapias, diagnóstico y políticas en salud.

