











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introducción
El síndrome de McCune-Albright (SMA) es una enfermedad genética rara, cuya prevalencia se estima aproximadamente entre 1/100 000 a 1/1 000 000 casos a nivel global. Se caracteriza por la triada clínica de displasia fibrosa poliostótica (DFP), pubertad precoz y manchas café con leche1.
La etiología molecular está asociada a la mutación post cigótica del gen GNAS que codifica la subunidad alfa de la proteína reguladora Gs. Esto conlleva a un aumento intracelular del adenosín monofosfato cíclico (AMPc) que resulta en el incremento de producción de melanina, estradiol, testosterona, tiroxina, hormona de crecimiento y cortisol. Generando cuadros como las manchas cafés con leche, pubertad precoz, acromegalia, hipertiroidismo y síndrome de cushing1,2. Además de ello, el aumento de AMPc en los osteoblastos genera una diferenciación a células estromales, causando DFP3.
La expresión de estos posibles cuadros clínicos, depende del tejido en el cual se encuentra esta mutación. Entre ellos, la pubertad precoz es la endocrinopatía más frecuente en niñas con SMA. Las manchas cafés con leche frecuentemente son la manifestación más temprana de esta enfermedad2.
El diagnóstico generalmente es clínico. También se puede realizar pruebas genéticas para detectar la mutación del gen GNAS, pero dado el mosaicismo que se presenta en este cuadro, es probable la presencia de falsos negativos. Por otro lado, un resultado positivo contribuye poco al manejo clínico3,4.
A continuación, se describe el caso de una paciente con síndrome de McCune-Albright que desarrolló acromegalia debido a macroadenoma hipofisiario productor de hormona de crecimiento (GH).
Presentación de caso
Paciente mujer de 44 años, con antecedente de pubertad precoz periférica, quien presentó menarquia a los 6 años de edad, refiere que, a los 12 años, notó aparición de tumoraciones pétreas en región naso genianas y antebrazo con crecimiento progresivo. A los 27 años, por presentar episodios de polimenorrea y menorragias persistentes que no remitieron al manejo médico, fue sometida a histerectomía y ooforectomía bilateral. A los 29 años paciente notó aumento de la talla del calzado y anillos, asociado a incremento progresivo del volumen de manos y pies. Se hospitalizó en el servicio para completar estudios diagnósticos y terapéuticos, debido a la sospecha clínica de acromegalia.
Al examen físico, la paciente presentaba las siguientes funciones vitales: presión arterial: 120/70 mm hg, frecuencia cardiaca: 76 latidos por minuto, frecuencia respiratoria: 16 respiraciones por minuto y saturación de oxígeno: 94 % a la fracción de inspiración de oxígeno ambienta. Las medidas antropométricas fueron peso: 54 kg, talla: 150 cm y circunferencia abdominal: 60 cm. Se evidenció facies acromegálica, en regular estado de hidratación y nutrición, con presencia de manchas cafés con leche de 1cm por 1,5 cm con leche en antebrazo, abdomen, manos y pies, así como melanosis labial inferior (Figura 1).
En el examen musculoesquelético, se encontró prominencia frontal, prognatismo, aumento de volumen y engrosamiento de manos y pies, así como tumoraciones pétreas en ambos antebrazos y en la región paranasal izquierda. Las maniobras de tinnel y phalen fueron positivas.
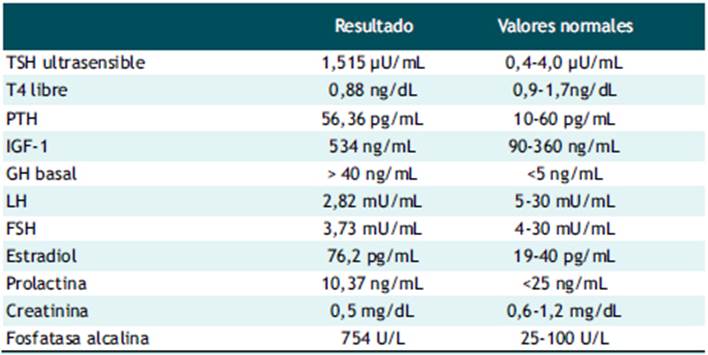
Dentro de los exámenes de sangre, la paciente presentó alteración del test de supresión de GH con 75 g de glucosa en todas las mediciones (> 1 ng/ml), siendo los valores de GH basal: 42 ng/ml, a los 60 minutos: 30,7 ng/ml, a los 120 minutos: 32,6 ng/ml (Tabla 1). El examen de survey óseo demostró la presencia lesiones expansivas múltiples tipo vidrio deslustrado de tipo benigno en todos los huesos del cuerpo compatible con displasia fibrosa poliostótica (Figura 2). La tomografía cerebral con contraste evidenció crecimiento del diploe en región parietal y temporal derecha. La tomografía de tórax con contraste mostró la presencia de imagen nodular de 5 mm en base de pulmón derecho (Figura 3). La resonancia magnética revela tumoración de adenohipófisis de crecimiento supraselar de 17x19x13 mm compatible con macroadenoma hipofisiario, que no comprime el quiasma óptico (Figura 4). Con el resultado de estos exámenes, el diagnóstico de la paciente fue acromegalia por macroadenoma hipofisiario, síndrome de Mccune-Albright, síndrome del túnel carpiano e hipogonadismo hipogonadotropo. La paciente fue sometida a resección transfenoidal del macroadenoma, evidenciándose 7 meses posterior a la cirugía, la presencia de remanentes del adenoma hipofisiario, asociado a un valor de factor de crecimiento insulínico tipo 1 (igf-1) alto (482ng/ml), compatible con persistencia de la enfermedad, por lo que actualmente recibe, manejo médico con cabergolina 1.5 mg semanal y permanece siendo evaluada por el servicio de endocrinología de nuestro hospital.
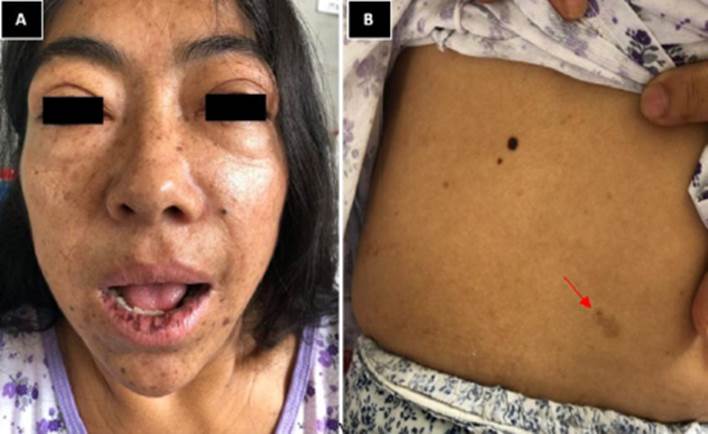
Figura 1 Figura 1. A) facies acromegálica: prominencia frontal, incremento de tejidos blandos, prognatismo. B) flecha apuntando a manchas de color café con leche a nivel del abdomen.
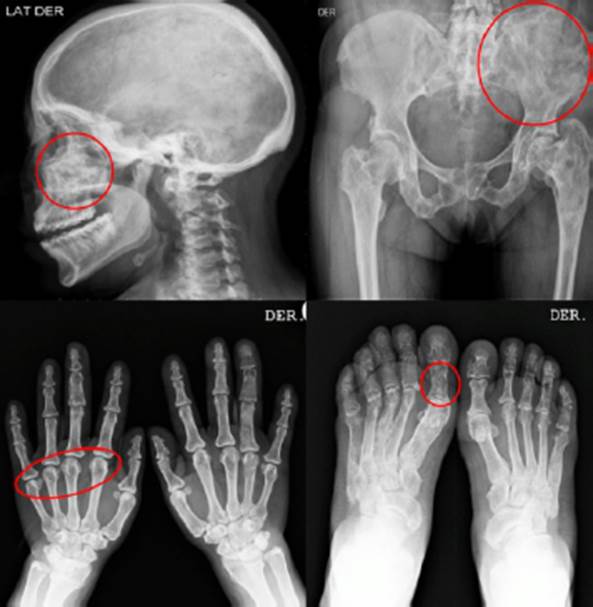
Figura 2 Survey óseo: displasia fibrosa poliostótica. Presencia de múltiples lesiones expansivas tipo "vidrio deslustrado" que no comprometen la región cortical, a nivel de cráneo, pelvis y extremidades.
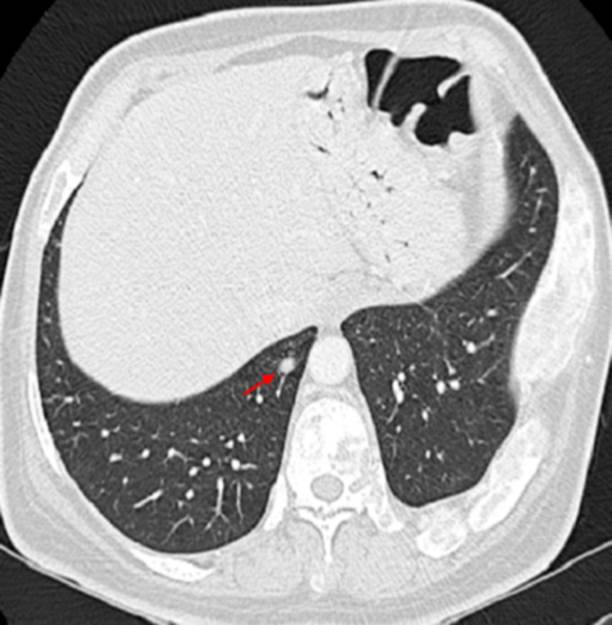
Figura 3 Tomografía axial computarizada de tórax. Flecha indica el nódulo de 5 mm en base de pulmón derecho.
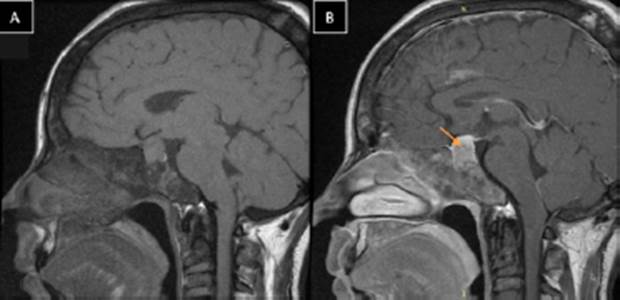
Figura 4 Resonancia magnética nuclear de hipófisis, flecha indica el macroadenoma hipofisiario. A. Sin contraste. B. Con contraste.
Discusión
En el Perú no existen estudios sobre la prevalencia del SMA, ni reportes de casos previos de la asociación de acromegalia con el mismo. En el año 1999, augusto et al. Reportaron el caso de un adolescente norteamericano con pubertad precoz, manchas cafés con leche, DFP y gigantismo5. A la actualidad, el presente caso es el primer reporte de una mujer con SMA y acromegalia en el Perú.
El SMA se encuentran asociado a endocrinopatías como pubertad precoz, enfermedad tiroidea, acromegalia y síndrome de Cushing1,2. Entre ellas, las más frecuente es la pubertad precoz periférica que forma parte de la triada clínica de esta patología3. Esto es producto de la activación autónoma de los ovarios generando así un incremento de los niveles de estradiol que conlleva a la clínica que presentó nuestra paciente, formación de quistes ováricos e inicio temprano de sangrado vaginal, asociado o no a desarrollo de mamas, crecimiento y maduración ósea6.
La segunda endocrinopatía más frecuente asociada al SMA es la enfermedad tiroidea7. Las características tiroideas en mas incluyen heterogeneidad localizada o difusa, lesiones quísticas y/o nodulares, junto con anomalías funcionales. Si bien la patología es más a menudo difusa, rara vez se asocia con clínica compresiva. Las anomalías funcionales, que rara vez se observan en ausencia de cambios ecográficos, se caracterizan por una función autónoma, con un aumento en la conversión intratiroidea de t4 hacia t3. Aunque se ha descrito cáncer de tiroides en pacientes con SMA, la prevalencia de no parece ser alta8. La afección tiroidea no estuvo presente en la paciente.
Por otro lado, en el presente caso, se evidencian manifestaciones clínicas y laboratoriales de acromegalia. Esta enfermedad presenta una prevalencia aproximada del 20 a 30 % en el SMA2,6. Las manifestaciones clínicas presentes son crecimiento desproporcionado de pies y manos, deformidad facial, perdida de la visión, audición u olfación, intolerancia a la glucosa, pudiendo también presentarse compromiso cardiaco. En presencia de displasia fibrosa craneofacial, aumenta la morbilidad y está asociada a alta prevalencia de macrocefalia, pérdida de la audición y neuropatía óptica9.
La acromegalia usualmente se asocia DFP del cráneo, especialmente de huesos de la base del cráneo10. La DFP es el componente más común del SMA. La afectación ósea es más frecuente en huesos del cráneo, principalmente la base del cráneo y el esqueleto axial, pudiendo haber una afectación de la visión y audición, es por ello que se recomienda tamizaje al momento del diagnóstico y en el seguimiento11. Las imágenes ayudan a evaluar la carga de enfermedad, siendo útil las radiografías, sin embargo, no ayudan a evaluar microfracturas o DFP incipiente, es por ello que la tomografía o resonancia magnética brindan una mejor evaluación del esqueleto óseo en lesiones incipientes12.
El exceso de hormona de crecimiento usualmente suele identificarse en el rango de edad de 3-64 años, con una edad media de 24,4, a diferencia de los pacientes con acromegalia sin SMA. En nuestra paciente el diagnóstico de acromegalia fue tardío, lo cual puede ser frecuente debido a que las deformidades esqueléticas que ocurren como resultado de la displasia fibrosa en SMA pueden enmascarar los cambios craneofaciales y acrales que se observan en la acromegalia 10. El impacto clínico de este retraso es significativo en el crecimiento tumoral, ya que 2/3 de los pacientes en el momento del diagnóstico tienen un macroadenoma hipofisario; desarrollo de complicaciones irreversibles como artropatía y apnea del sueño; así como aumento de la mortalidad13. El diagnóstico confirmatorio se realiza por medio de los niveles de GH tras el test de supresión con glucosa, así como por los niveles elevados de IGF-1 según edad14.
Adicionalmente el hipogonadismo hipogonadotrópico evidenciable que presenta la paciente, el cual es frecuente en la acromegalia, se debe a la destrucción de las células secretoras de GnRH, propia del efecto compresivo del macroadenoma hipofisiario15.
Existen varias modalidades del tratamiento de la acromegalia: quirúrgico, farmacológico (análogos de somatostatina, cabergolina y pegvisomant) y radioterapia14. La resección quirúrgica del adenoma hipofisiario generalmente es la primera opción de tratamiento, sin embargo, se reporta que la tasa de curación es baja en comparación con pacientes con acromegalia sin SMA16. Adicionalmente la displasia fibrosa del hueso esfenoides, que no se observó en nuestra paciente, puede impedir un abordaje transesfenoidal exitoso de la silla turca. Por estas razones, la mayoría de los pacientes con SMA no suelen ser candidatos de cirugía hipofisaria17.
En el caso de la paciente, el tratamiento quirúrgico fue el tratamiento inicial, sin embargo, en el postoperatorio, al evidenciarse persistencia de la enfermedad, se optó por el manejo médico con agonistas dopaminérgicos, siendo el manejo médico del exceso de GH e IGF-1 de suma importancia para los pacientes con acromegalia asociada a SMA.
Conclusión
El síndrome de McCune Albright es una enfermedad infrecuente, en el cual pueden verse comprometidos múltiples ejes endocrinos, entre ellos el eje de la hormona de crecimiento, pudiendo asociarse a acromegalia. El diagnóstico y tratamiento oportuno de esta enfermedad y sus complicaciones asociadas permitirá un mejor pronóstico en los pacientes con SMA.

