











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
Las histiocitosis son enfermedades raras y a menudo sistémicas, caracterizadas por la infiltración tisular de histiocitos anormales con características morfológicas e inmunohistoquímicas peculiares. 1 Se desconocen los mecanismos exactos que producen las histiocitosis; sin embargo, algunas son trastornos clonales, a menudo impulsados por mutaciones somáticas de protooncogenes, mientras que otras pueden ser reactivas o estar asociadas a enfermedades sistémicas inmunomediadas o infecciosas. 1,2 Con base a sus características clínicas, histológicas, fenotípicas, genéticas y/o moleculares, las histiocitosis se dividen en cinco grupos, a conocerse: 1) grupo de células de Langerhans (HCL) (Grupo L); 2) histiocitosis de células no-Langerhans (HNL) cutáneas y mucocutáneas (Grupo C); 3) enfermedad de Rosai-Dorfman (Grupo R); 4) histiocitosis malignas (Grupo M) y; 5) linfohistiocitosis hemofagocítica y síndromes de activación de los macrófagos (Grupo H). 3
Actualmente se conoce que las histiocitosis se presentan a cualquier edad; empero, son más frecuentes en menores de 18 años (67% de los casos). 4 La HCL tiene una incidencia de 4,6 casos/millón en niños menores de 15 años, y de 1-2 casos/millón en pacientes mayores de 15 años, con un ligero predominio del sexo masculino (relación de 1.2-1.5). 5,6 La histiocitosis de células de Langerhans (HCL) es la forma de presentación más frecuente. Mientras que, de las HNL, el xantogranuloma es el más frecuente; mismo que suele producirse en el primer año de vida con desaparición espontánea (forma juvenil) y en individuos de mediana edad sin remisión espontánea (forma adulta). Por otro lado, la enfermedad de Erdheim-Chester (ECD) suele aparecer entre los 40 y los 70 años de edad y presenta una predominancia masculina del 73%. 5 La combinación de las HCL y las HNL es fortuita y muy rara. 6 Según el Registro Nacional de Tumores del Instituto Nacional de Lucha Contra el Cáncer (SOLCA), la incidencia de la histiocitosis es muy baja en el país. 7
El objetivo de este manuscrito es reportar el caso de una histiocitosis mixta en una paciente de 66 años de edad, atendida en el Hospital de Especialidades “José Carrasco Arteaga” (IESS), Cuenca, Ecuador. Para ello, se siguieron los lineamientos CARE (Case Report Guildness, por sus siglas en inglés) para el reporte de casos clínicos.
REPORTE DE CASO
Paciente femenina de 66 años de edad, procedente y residente de Guayaquil-Guayas, enfermera, jubilada por invalidez. Fumadora desde los 16 años de edad, suspende consumo de tabaco a partir de 2018. Con antecedentes personales patológicos de hipotiroidismo, diabetes insípida y gastropatía crónica. Entre sus antecedentes familiares patológicos destacan diabetes mellitus tipo 2 e hipertensión arterial maternas.
Refiere, desde hace cuatro años, presencia de masas tumorales, no dolorosas, localizadas en la cara anterior de las piernas, dos en ellas en la pierna izquierda y dos en la pierna derecha; de crecimiento progresivo, pruriginosas, con vasculatura visible e importante aumento del volumen pretibial, bilateral, con predominio derecho. Se niega la presencia de secreción purulenta, sanguinolenta u otros. Evaluaciones anteriores las catalogaron como tumores benignos lipomatosos y/o hematomas. Sin tratamiento para la actualidad. También, paciente refiere presencia de zona alopécica en región occipital; diagnosticada como psoriasis por la presencia de placas y descamación superficial; recibiendo tratamiento con base a corticosteroides, sin mejoría alguna. Además, el familiar menciona cuadros de otitis media a repetición (aproximadamente 9 episodios), desde hace cuatro años, con importante inflamación de la región retroauricular. Finalmente, refiere dolor óseo generalizado, de tipo crónico, de tres años de evolución, que desde hace un mes se exacerba, con intensidad 6/10 (EVA), de tipo compresivo, que no cede a analgésicos, y afecta la movilidad, llegando a necesitar ayuda de otra persona. Concomitantemente, refiere fiebre durante las noches, de 39-40 °C; epigastralgia, náuseas, disnea de esfuerzos moderados, fatiga, debilidad generalizada, sudoración nocturna y pérdida de peso de 5 kg en los últimos 2 años.
Al examen físico, en cuero cabelludo: placas grisáceas descamativas y nódulos blanquecinos que rodean los folículos pilosos. En ojos: conjuntivas palpebrales pálidas y enoftalmos. En boca: mucosas semihúmedas, ausencia de piezas dentarias, empleo de prótesis. En cuello: ganglios retroauriculares, bilaterales, no dolorosos, fijos a la palpación, el mayor de 1.5x1 cm. En piel: disminución de la turgencia, resequedad y palidez generalizada. En miembros inferiores: presencia de nódulos tumorales en cara anterior de las piernas, con telangiectasias superficiales, calientes al tacto, duras, poco móviles, con presencia de placas eritematocostrosas en superficie, pruriginosas, con signos de rascado y xerodermia. En pierna izquierda: masa superior de 4x4 cm e inferior de 1x1 cm. En pierna derecha: masa superior de 3x4 cm e inferior de 2x1 cm. (Figura 1, A.)
En la analítica de ingreso, la paciente presentó leucocitosis (15’120/uL) con neutrofilia (10’970/uL) y monocitosis (1’340/uL), trombocitosis reactiva (671’000/uL) con JAK2 negativa, anemia microcítica hipocrómica (Hb 10.1 g/L, VCM 71 ft, MHC 23.2 pg). Tiempos de coagulación prolongados (TTP 33.7 seg, fibrinógeno 528 mg/dL). Función tiroidea alterada (TSH 5.30 mUI/L, T3 4.27 pg/mL, T4 1.0 ng/dL), pese a tratamiento para hipotiroidismo. Aumento de la lambda (907 mg/dL), B2 microglobulina (4888 ng/mL) e IgG (1610 mg/dL), sentando la probabilidad de un proceso maligno o neoplásico en curso.
Con estos antecedentes y la sospecha de compromiso sistémico, se realizó una serie ósea metastásica, en la que se visibilizó disminución generalizada de la densidad ósea y presencia de lesiones blásticas y líticas a diferentes niveles. (Figura 1, B.) También, se realizó una tomografía computarizada simple y contrastada de cabeza, tórax, abdomen y pelvis; en la que se evidenció: sinusitis maxilar crónica y pérdida de la señal normal de la neurohipófisis, nódulos reticulonodulares y quistes de ocupación multilobular bilateral, derrame pericárdico, lesiones óseas múltiples, quiste hepático, enfermedad inflamatoria renal bilateral con quistes medulares bilaterales y aspecto de pelos perirrenales, y lesión suprarrenal izquierda de probable origen metastásico. (Figura 1, C.) Además, una endoscopia y biopsia gástrica se consideraron, mostrando metaplasia intestinal completa del antro. El ecocardiograma mostró disfunción diastólica del ventrículo izquierdo grado I. La espirometría descartó enfermedad pulmonar obstructiva crónica.
Con lo anterior, se sospechó de una probable histiocitosis de células de Langerhans y una histiocitosis de células no Langerhans o enfermedad de Erdheim Chester. (Tabla No. 1) Por tal motivo, se solicitó biopsia de placas a nivel del cuero cabelludo y biopsia escisional de masas tumorales en pierna derecha, reportándose una proliferación de tipo histiocítica, compuesta por dos poblaciones bien diferenciadas. Posteriormente, se envía muestras a inmunohistoquímica, encontrándose positividad para CD68, fascina, S100 y CD1a. La sinaptofisina, CD163, CD23, CD21 y otros marcadores linfoides no estuvieron disponibles. (Figura 1, D.)
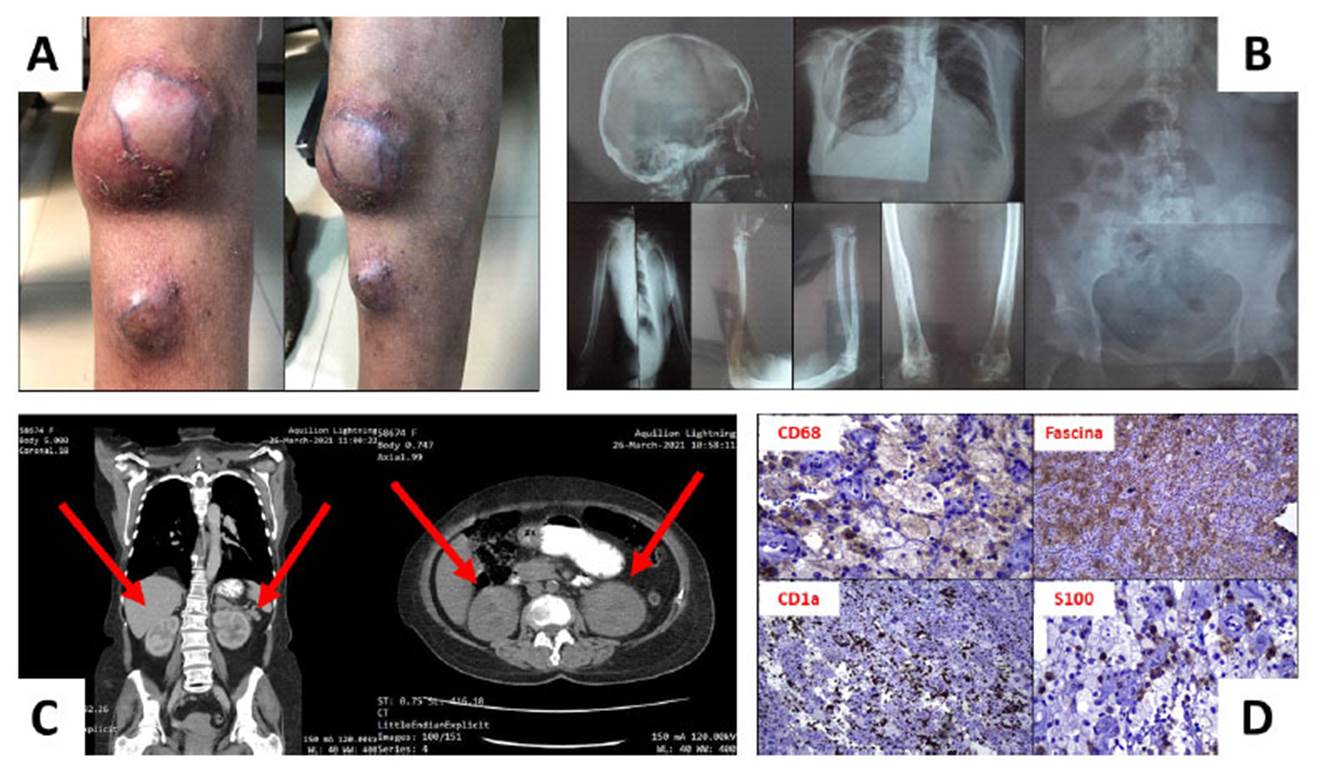
Figura 1 Manifestaciones clínicas, imagenológicas e inmunohistoquímicas encontradas: A) Nódulos tumorales en cara anterior de pierna izquierda; se evidencia xerosis cutis y señales de rascado [Cuenca, Azuay, Ecuador; 08/04/2021]. B) Serie ósea metastásica en la que se muestra disminución generalizada de la densidad ósea, presencia de múltiples lesiones blásticas en cráneo, engrosamiento cortical a nivel del tercio medio de la diáfisis humeral, imágenes líticas que condicionan adelgazamiento de la cortical en cabeza, cuello y cóndilos femorales, región metaepifisiaria proximal de la tibia izquierda, cúbito y radio; no estuvieron disponibles las tomas laterales de columna [Servicio de Imagenología y Radiología del Hospital “José Carrasco Arteaga”. Cuenca, Azuay, Ecuador; 16/03/2021]. C) Tomografía computarizada simple y contrastada. A nivel de tórax, opacidades bilaterales con patrón reticulonodular simétrico y lesiones quísticas de predominio en lóbulos superiores, segmento medial del lóbulo medio derecho y porción superior y media de los lóbulos inferiores. Además, placas de ateroma calcificadas en aorta ascendente y cayado aórtico, presencia de derrame pericárdico moderado, T8 plana y lesiones permeativas en el cuerpo vertebral de T3, T5, T6, T9 y T11. A nivel de abdomen, imagen hipodensa ovalada con bordes definidos en el segmento IV del hígado, riñones con caliectasia bilateral así como engrosamiento capsular bilateral, densa infiltración de la grasa perirrenal dando el aspecto de riñones pelúcidos, quistes medulares, al menos dos en riñón derecho, el mayor de 23x14 mm y cuatro en el izquierdo, el mayor de 22.5x16.1 mm. Adicionalmente, glándula suprarrenal izquierda engrosada irregularmente con pérdida de su morfología, ateromatosis aorto-ilíaca, lesiones permeativas en cuerpo vertebral de vértebras lumbares y las dos primeras sacras, así como en las alas iliacas, ramas públicas, alas sacras y en lo valorable del fémur lateral [Servicio de Imagenología y Radiología del Hospital Municipal de Cuenca. Cuenca, Azuay, Ecuador; 23/03/2021]. D) Pruebas de inmunohistoquímica positivas para: CD68, fascina, CD1a y S100 [Cuenca, Azuay, Ecuador; 23/03/2021].
Tabla 1 Diagnóstico diferencial entre las enfermedades histiocíticas más frecuentes.
| Diagnósticos diferenciales | Hallazgos clínicos y exámenes complementarios positivos | Hallazgos clínicos y exámenes complementarios negativos |
| Histiocitosis de células de Langerhans | Óseas: Lesiones osteolíticas en huesos largos. | Cardíacas: Raramente se reporta afectación a este nivel. |
| Endocrinas: Hipodensidad en la neurohipófisis. Diabetes insípida. | Retroperitoneales: Rara vez se reporta afectación renal y suprarrenal. | |
| Hipotiroidismo. | Vasculares: No es frecuente ver afectación arterial en estos pacientes. | |
| Respiratorias: Disnea. Nódulos reticulonodulares y quistes multilobulares (en etapas posteriores y relacionadas con el tabaquismo). | ||
| Dermatológicas: Masas tumorales subcutáneas en miembros inferiores. | ||
| Sistema reticuloendotelial y hematopoyéticas: Afectación hepática (quistes hepáticos). | ||
| Linfáticas: Adenopatías retroauriculares. | ||
| Histopatología: Identificación de población histiocítica en muestra de biopsia. CD68 +, s100 +, CD1a +. | Dermatológicas y linfáticas: No es frecuente la presencia de masas tumorales ni adenopatías. | |
| Histiocitosis de células no Langerhans o Enfermedad de Erdheim Chester | Óseas: Lesiones osteo-escleróticas en huesos largos (diáfisis) y en el cráneo. | Histopatología: No es frecuente que haya positividad en el marcador tumoral CD1a. |
| Endócrinas: Hipodensidad en la neurohipófisis. Diabetes insípida. | ||
| Hipotiroidismo. | ||
| Respiratorias: Engrosamiento del seno maxilar. | ||
| Cardíacas: Derrame pericárdico. | ||
| Arteriales: Infiltración periaórtica o “aorta recubierta”. | ||
| Sistema reticuloendotelial y hematopoyético: Afectación hepática (quistes hepáticos). | ||
| Retroperitoneales (renal): Infiltración perirrenal o “riñones peludos”, con extensión a las glándulas suprarrenales. | ||
| Histopatología: Identificación de población histiocítica en muestra de biopsia. CD68 +, S100 -/+. |
Finalmente, se estableció el diagnóstico de histiocitosis mixta con compromiso sistémico. Se recomendó realizar estudios BRAF V600E para determinar la idoneidad del tratamiento. Sin embargo, estos no estuvieron disponibles. Dentro de las terapias dirigidas contra la enfermedad, se sugirieron: anticuerpos monoclonales anti BRAF V600E, interferón alfa (INFa) y/o quimioterapia. Siendo que, el pronóstico será favorable siempre que se instaure tratamiento. La paciente solicitó ser tratada en su lugar de origen, sin embargo, rechazó el tratamiento; y falleció días después. Motivo por el que no se pudo realizar un seguimiento del caso.
DISCUSIÓN
Las histiocitosis son enfermedades heterogéneas que comprenden la histiocitosis de células de Langerhans y la enfermedad de Edhereim Chester, principalmente. 9,10 Estas son entidades poco frecuentes, con características clínicas, radiológicas, histológicas y moleculares propias. Sin embargo, su coexistencia es excepcional; siendo que, hasta la actualidad, se han reportado 105 casos a nivel mundial, de los que, 75 casos corresponden a pacientes adultos. 8 Se han descrito tres tipos de histiocitosis mixta, de las cuales, la histiocitosis mixta tipo 1 es la más frecuente. 8,9 Este es, probablemente, el primer reporte de caso de histiocitosis mixta publicado en Ecuador, pese a que la incidencia de la enfermedad es baja en el medio y aún desconocida a nivel de Latinoamérica. 7,8
La paciente en mención, presentó hallazgos clínicos y radiológicos similares a los descritos en la literatura. Las manifestaciones clínicas más tempranas fueron las de HCL en comparación con las de EDC. 8,9 La paciente presentó afectación multisistémica, pero los hallazgos clínicos-imagenológicos más orientadores para el diagnóstico de histiocitosis mixta fueron: el antecedente de tabaquismo de larga data, las lesiones dermatológicas (HCL), las alteraciones endocrinológicas concomitantes (HCL/EDC), las lesiones óseas líticas (HCL) y escleróticas (EDC) y la infiltración perineal con aspecto de “riñones pelúcidos” (EDC), además de las lesiones arteriales periaórticas y los signos de derrame pericárdico (EDC). 9,10 Dentro del espectro de manifestaciones clínicas también se evidenció en la paciente, gastropatía crónica con hallazgos endoscópicos de metaplasia intestinal completa del antro y la presencia de un quiste hepático en la tomografía axial computarizada de abdomen, los cuales no han sido reportados como un componente de histiocitosis mixta en la actualidad. Probablemente, son consecuencia de patologías concomitantes. La biopsia incisional de las lesiones, con posterior estudio histopatológico e inmunohistoquímico, fueron esenciales para el diagnóstico definitivo. Pese a no disponer de todos los marcadores moleculares, se encontraron en esta paciente patrones moleculares de HCL y EDC con infiltrado histiocítico bien definido y positividad para los marcadores CD68+/CD1a/S100+ y CD68+/S100+, respectivamente. 9,10
Según el Mayo Clinic Histiocytosis Working Group Consensus Statement for the Diagnosis and Evaluation of Adult Patients with Histiocytic Neoplasms, el gold standard para el diagnóstico de la HCL y EDC es el estudio histopatológico en un contexto clínico y radiológico sugerente. 10 Las pruebas de inmunohistoquímica son complementarias y permiten la identificación y clasificación de las enfermedades histiocíticas. De tal forma, en la HCL, las células de Langerhans tienen núcleos ligeramente agrandados, con surcos nucleares delicados y una cromatina menos condensada. El análisis inmunohistoquímico muestra un fenotipo con expresión de S100, CD1a y langerina. 10 Por otro lado, la EDC se caracteriza por acumulaciones de histiocitos típicamente espumosos o cargados de lípidos y células gigantes de Touton en un fondo fibrótico. 10,11) Los histiocitos muestran marcadores positivos, como CD68, CD163 y el factor XIIIa; y son negativos para CD1a y langerina. 10 En raras ocasiones se ha observado positividad para S100. 5 Además, las mutaciones BRAF V600E (EDC 50% vs. HCL 55%) y MAP2K1 (EDC 18% vs. HCL 55%) pueden estar presentes en mayor o menor medida y contribuir al diagnóstico. 5 En nuestro caso, el hallazgo de células histiocíticas y la positividad para los marcadores CD68+, CD1a y S100+, a más de la clínica con la que debutó la paciente, permitieron realizar el diagnóstico de histiocitosis mixta.
Respecto a la mutación BRAF V600E y MAP2K1, vale mencionar que estas son clave para instaurar la terapia dirigida más idónea para el paciente, sumado al compromiso sistémico y la gravedad de la enfermedad. Entre estos, están los inhibidores BRAF (p.ej.: emurafenib, dabrafenib, encorafenib) y los inhibidores de la vía MAP2K/ERK (p.ej.: cobimetinib, trametinib, binimetinib, selumetinib). 11 Otras terapias con base a citosinas, citotóxicos (p.ej.: cladribina, ciclofosfamida, vinblastina y metotrexato), interferón-a-2a (IFN-a-2a) e IFN-a-2a pegilado son también apropiadas y aún son una posibilidad para el tratamiento. 5,11
Las principales limitaciones en el abordaje de la paciente fueron la falta de disponibilidad de pruebas moleculares e inmunohistoquímicas como la prueba de mutaciones del gen BRAF V600E, mutaciones del gen MAP2K1 y la langerina que presentan alta sensibilidad en pacientes con EDC e HCL, respectivamente. Además, estudios imagenológicos sensibles y específicos como la tomografía por emisión de positrones, resonancia magnética nuclear y/o la gammagrafía ósea hubieran sido de gran utilidad. Pues, estas permiten identificar y caracterizar lesiones óseas muy sutiles (Tabla 1), identificar el lugar más adecuado para realizar la biopsia y tomar acciones para prevenir posibles fracturas. 9,10 Por tanto, estos estudios hubieran permitido confirmar más rápido y con mayor precisión el diagnóstico de histiocitosis mixta, y a su vez, establecer lineamientos claros respecto a la terapéutica.
CONCLUSIÓN
Es posible realizar el diagnóstico de histiocitosis a partir de los antecedentes personales patológicos del paciente y los hallazgos clínicos manifiestos, con el apoyo de estudios radiológicos, histológicos e inmunohistoquímicos sensibles y específicos. Por último, las características de la enfermedad de Edhereim Chester y la histiocitosis de células de Langerhans pueden llegar a solaparse

