











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
El síndrome de Noonan es un trastorno genético y es uno de los síndromes no cromosómicos más frecuentes1. Una variación en el gen PTPN11 en el locus 12q24 podría explicar el fenotipo en la mayoría de las personas con síndrome de Noonan2. Este gen participa en la vía de las proteínas quinasas activadas por mitógeno (RAS -MAPK), que está implicada en la proliferación, migración, diferenciación y oncogénesis celular3.
Los síndromes con una mutación somática en algún gen que participa en la vía RAS-MAPK son llamadas enfermedades neuro-facio-cardio-cutáneas o RASopatias4. Estos incluyen el síndrome de Noonan, el síndrome Noonan-like con cabello anágeno caduco, síndrome de Noonan-like con o sin leucemia mielomonocítica juvenil, síndrome de Noonan con múltiples lentigos, síndrome-cardio facio-cutáneo, entre otros5. La presentación clínica entre estas enfermedades se superpone, lo que implica un desafío diagnóstico6.
La identificación de mutaciones somáticas en los genes implicados en la vía RAS-MAPK apoya el diagnóstico de las RASopatias. Del mismo modo, un paciente con síndrome de Noonan sin mutación del gen PTPN11 es generalmente positivo a una mutación en los genes SOS1, RAF1, KRAS, NRAS, MAP2K1, BRAF, o CBL7. Sin embargo, el resto de las RASopatias están también caracterizadas por la mutación de algún gen específico mencionado anteriormente5. Esta situación representa una superposición adicional en las mutaciones genéticas, lo que dificulta aún más el diagnóstico.
Presentamos el caso de un recién nacido con sospecha clínica de síndrome de Noonan desde el embarazo. El diagnóstico se confirmó durante el seguimiento postnatal, donde cumplió con los criterios clínicos de diagnóstico. Adicionalmente, se identificó la concurrencia de una mutación somática de novo en el gen MAP2K1, una variación del gen CBL de significado incierto y la negatividad para mutaciones del gen PTPN11. Seguimos las pautas de la guía CARE para reporte de casos8.
REPORTE DE CASO
Recién nacido varón de 34 semanas de edad gestacional nacido por cesárea. Su madre es una mujer de 31 años de edad, quien presentó una infección urinaria a los cinco meses de embarazo con tratamiento antibiótico completo. Presenta hipertensión desde la adolescencia por lo que se medica con alfa-metildopa diariamente. Durante el embarazo, una ecografía gestacional a las 24 semanas mostró hidronefrosis, higroma quístico y polihidramnios de 22,9 mm3. Los resultados de la amniocentesis mostraron un cariotipo 46, XY normal. A las 32 semanas de gestación otra ecografía mostró hidronefrosis bilateral con polihidramnios persistente. A las 34 semanas de gestación, el polihidramnios empeoró a 463 mm3 (Figura 1). En este punto, la madre fue sometida a una cesárea de emergencia.
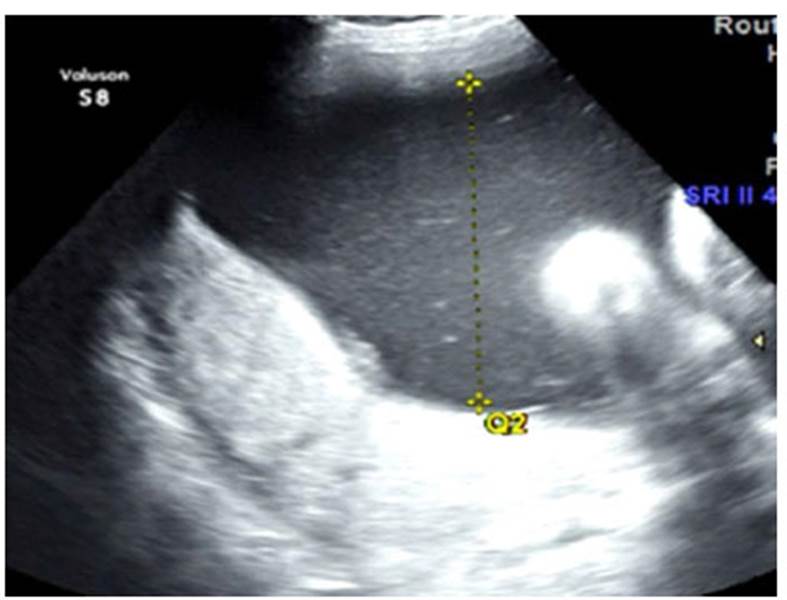
Figura 1 Ecografía a las 34 semanas de gestación que muestra hidronefrosis bilateral, polihidramnios grave con un índice de líquido amniótico de 404 mm. El estudio Doppler no mostró alteración de las arterias uterinas ni signos de cambios hemodinámicos compensatorios fetales.
El recién nacido pesó 3030 gramos, con talla de 46 cm y circunferencia cefálica de 32 cm. (Percentiles de peso, talla y perímetro cefálico según la tabla de crecimiento de Olsen para bebés prematuros y a término para niños 9: Percentil >97 °, 50 ° y 75 °, respectivamente). Presentó signos vitales, temperatura y APGAR normales. En el examen físico, se observó edema moderado a severo en la nuca y en ambos párpados, además de cejas escasas, puente nasal ancho, hendiduras palpebrales descendentes y pabellón auricular de implantación baja. El murmullo vesicular se encontró ligeramente disminuido en ambos hemitórax, se auscultó un murmullo sistólico de intensidad II/VI. Además, presentó criptorquidia derecha y tono muscular ligeramente disminuido. El resto del examen físico fue normal.
En los primeros 30 minutos de nacimiento presentó dificultad respiratoria. Luego, fue transferido a la unidad de cuidados intermedios de neonatología, con soporte de oxígeno donde recibió surfactante pulmonar. El pediatra ingresó al recién nacido a hospitalización con los siguientes diagnósticos: Recién nacido prematuro grande para la edad gestacional, síndrome de dificultad respiratoria, síndrome dismórfico, higroma cervical posterior, hidronefrosis y un posible defecto cardíaco congénito acianótico.
La radiografía de tórax al nacimiento y en la primera semana después del nacimiento mostró una acentuación de la trama bronco-vascular, con ninguna otra alteración. La ecocardiografía evidenció un defecto del tabique auricular ostium secundum de 9 mm., con una derivación de izquierda a derecha, ausencia de vena cava inferior, hipertensión pulmonar leve (35 mmHg.) y persistencia del conducto arterioso de 1,4 mm (Figura 2). La ecografía renal confirmó la hidronefrosis bilateral. El proteinograma en orina mostró 12 mg/dL de albúmina, 4 mg/dL de alfa-2-globulina, 2 mg/dL de beta-globulina y 1 mg/dL de gamma-globulina
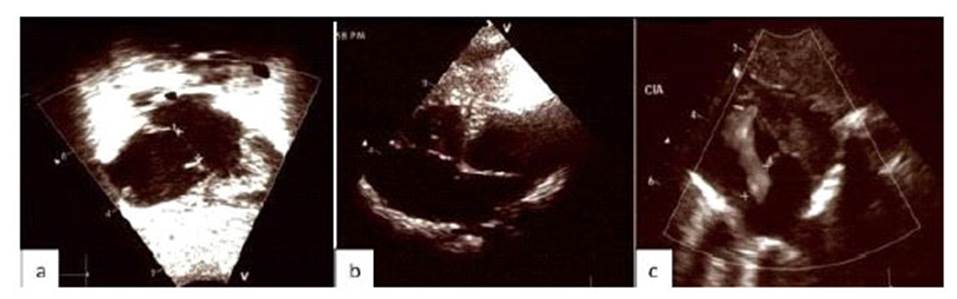
Figura 2 2a. Ecocardiografía a las 24 horas después del nacimiento, muestra el defecto septal auricular ostium secundum de 9 mm, con una derivación de izquierda a derecha, ausencia de vena cava inferior e hipertensión pulmonar leve. Figura 2b: Ecocardiografía 1 semana después del nacimiento muestra el defecto septal auricular extenso de 12 mm, sin cambios adicionales en comparación con el anterior, y sin hipertensión pulmonar. Figura 2c: Ecocardiografía a las 3 semanas después del nacimiento, muestra la comunicación interauricular de 10 mm, isomerismo auricular izquierdo y ausencia de hipertensión pulmonar.
La tomografía del abdomen y pelvis mostró hidronefrosis bilateral moderada con parénquima renal simétrico y preservado. El resto de los órganos abdominales y pélvicos no presentaron cambios relevantes (Figura 3). El recuento sanguíneo, los niveles séricos de proteína C reactiva, el análisis de orina completo y el cultivo de orina no mostraron alteraciones significativas. En la evaluación por oftalmología y neurología pediátrica se diagnosticó retina inmadura e hipotonía moderada, respectivamente
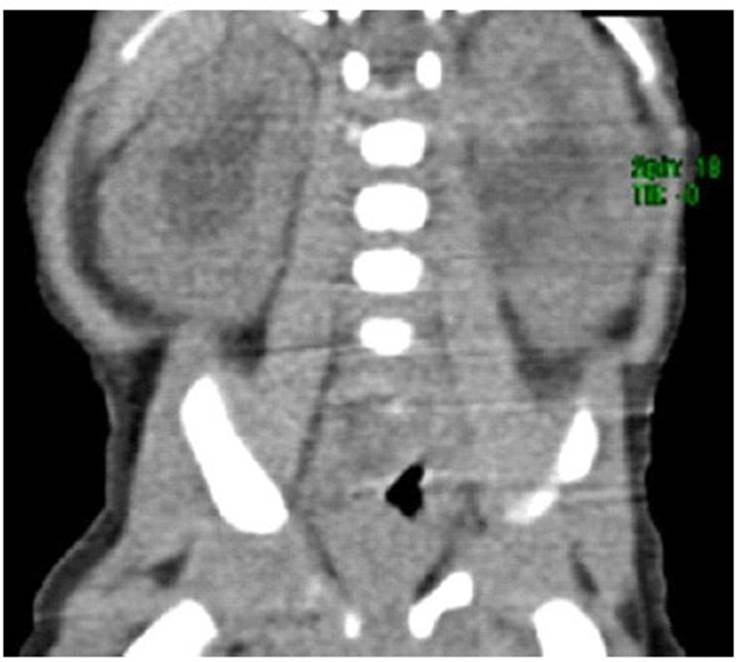
Figura 3 Tomografía espiral multicorte abdominal y de pelvis: Hidronefrosis moderada bilateral. Parénquima renal simétrico con espesor normal. Órganos abdominales retroperitoneales sin cambios relevantes.
Ante la sospecha genética de síndrome de Noonan. Se envió un panel de 14 genes asociado al síndrome de Noonan a los Laboratorios Invitae (California, Estados Unidos) para confirmar el diagnóstico. Se identificó una variante patógena en el MAP2K1 que consistía en un cambio de adenina a guanina en la posición 199 c.DNA en el gen MAP2K1. Además, identificó una variante de significancia incierta en el gen CBL que consistía en un cambio de citocina a timina en la posición 2345 c.DNA en el gen CBL. Las pruebas parentales revelaron que los padres no eran portadores de la mutación en el gen MAP2K1 ni en el gen CBL.
El paciente tuvo una evolución favorable durante la hospitalización. El paciente fue dado de alta a casa con leche materna y/o fórmula hidrolizada a demanda, tratamiento con hidroclorotiazida y espironolactona.
Posteriormente, diferentes especialidades pediátricas realizaron seguimientos. Fue adicionalmente diagnosticado con retraso psicomotor, blefaroptosis de ojo izquierdo, enfermedad de reflujo gastroesofágico, y anemia ferropénica. Además, durante el control post natal en la semana 23 de vida, se identificó el percentil de talla < 2, adicionando el criterio mayor faltante para el diagnóstico definitivo (Tabla 1). Actualmente, todos sus diagnósticos son controlados.
Tabla 1 Seguimiento médico postnatal del paciente desde el alta médica a las 70 semanas luego del nacimiento.
| Edad postnatal (semanas) | Edad corregida (semanas) | Talla (cm) | Percentil de altura * | Departamento | Diagnóstico relevante o nuevo para el seguimiento | Manejo |
| 0 | 34 (edad gestacional) | 46 | 50 | Hospitalización | Síndrome de Noonan Prematuro, Hidronefrosis, Defecto del tabique auricular, Retina inmadura | Espironolactona 3 mg una vez al día Hidroclorotiazida 3 mg bid |
| 4 | 38 (edad gestacional) | 49 | 50 | Pediatría | Retina inmadura | Ninguno |
| 6,5 | 0.5 0.5 | NA | NA | Cardiología pediátrica | Defecto del tabique auricular, Displasia broncopulmonar | Espironolactona 3 mg una vez al día Hidroclorotiazida 3 mg bid |
| 8 | 2 | 53 | 50 | Pediatría | Displasia broncopulmonar | Antibiótico profiláctico: Cefadroxil 1 ml cada día durante 30 días. |
| 12 | 6 6 | 55 | 30 | Pediatría | Hipotonía congénita Anemia por deficiencia de hierro | Hierro polimaltosado 50 mg/ml: 10 gotas vía oral por día durante 30 días |
| 16,5 | 10,5 | NA | NA | Cardiología pediátrica | Defecto del tabique auricular | Ninguno |
| 19 | 13 | NA | NA | Oftalmología | Blefaroptosis del ojo izquierdo | Explicar a la madre los riesgos y beneficios del tratamiento quirúrgico. |
| 23 | 17 | 59 | <2 | Pediatría | ||
| 24 | 18 | 59 | <2 | Gastroenterología pediátrica | Enfermedad por reflujo gastroesofágico | Lanzoprazol 15 mg. ½ paquete con 15 ml. de agua, una vez al día |
| 33 | 27 | 64 | 2 | Pediatría | Anemia por deficiencia de hierro, enfermedad por reflujo gastroesofágico | Lanzoprazol 15 mg. ½ paquete con 15 ml. de agua, una vez al día Hierro polimaltosado 50 mg/ml: 10 gotas vía oral por día durante 30 días |
| 40 | 34 | 64 | <2 | Endocrinología pediátrica | Enfermedad por reflujo gastroesofágico | Ninguno |
| 45 | 39 | 66 | <2 | Pediatría | Constipación, anemia ferropénica, blefaroptosis, hidronefrosis, defecto del tabique auricular, retraso psicomotor | Hierro polimaltosado 50 mg/ml: 10 gotas vía oral por día durante 30 días Lactulosa 3 ml bid por 3 días |
| 47 | 41 | 66 | <2 | Gastroenterología pediátrica | Enfermedad por reflujo gastroesofágico, disfagia | Lanzoprazol 15 mg. 15 ml de agua, una vez al día |
| 60 | 54 | Oftalmología | Blefaroplastia del ojo izquierdo | |||
| 62 | 56 | 69 | <2 | Gastroenterología pediátrica | Enfermedad por reflujo gastroesofágico, disfagia | Lanzoprazol 15 mg. 15 ml de agua, una vez al día |
| 70 | 64 | 73 | <2 | Pediatría | Constipación Hidronefrosis, defecto del tabique auricular, retraso psicomotor | Lactulosa 4 ml bid por 3 días |
DISCUSIÓN
Reportamos un recién nacido con sospecha de síndrome de Noonan al nacer. Confirmamos el diagnóstico clínico durante el seguimiento postnatal identificando una mutación de novo en la línea germinal del gen MAP2K1 y una variante de significado incierto en el gen de la CBL, con gen PTPN11 normal. Destacamos la importancia de la sospecha inicial de esta enfermedad y la siguiente búsqueda con paneles genéticos asociados, que pueden preceder al cumplimiento completo de los criterios clínicos.
El síndrome de Noonan puede ocurrir hasta en uno de cada 1000 o 2500 nacimientos vivos10. La sucesión familiar sigue a una herencia autosómica dominante. Sin embargo, las mutaciones de novo ocurren en el 60% de los casos, como en el presente paciente2. No existe una predilección específica conocida por raza o sexo11. El síndrome de Noonan se caracteriza por una gran variedad de hallazgos clínicos, lo que dificulta la identificación de individuos ligeramente afectados. Para este propósito, van der Burgt propuso criterios de diagnóstico12. El presente caso inicialmente no cumplía con los criterios completos, sólo los criterios menores: 1) dismorfología sugestiva en rostro 2) criptorquidia, y 3) otros defectos cardíacos. Es durante el periodo de seguimiento, cuando finalmente cumple con el criterio mayor de percentil de talla < 3, realizando finalmente el diagnóstico definitivo.
Anteriormente, algunos autores propusieron correlaciones genotipo-fenotipo en pacientes con síndrome de Noonan6,13. Las características faciales de los pacientes con síndrome de Noonan generalmente se acentúan con la edad14. Como se observó, durante el breve periodo de seguimiento del presente caso, sus rasgos faciales sugestivos, pero no típicos se mantuvieron similares. Nuestro paciente presenta signos faciales leves como edema nucal y hendiduras palpebrales descendentes, que son característicos en pacientes de América Latina15, además de cejas escasas, puente nasal ancho y orejas de implantación baja, que son características en pacientes con mutación del gen MAP2K116.
Además, el paciente presentaba anomalías congénitas cardiacas, urinarias, oftalmológicas, y gastrointestinales significativas. En el sistema cardiovascular, el paciente presentaba dos anomalías. La comunicación interauricular es una de las anomalías más frecuentes encontradas en pacientes con síndrome de Noonan, y el conducto arterioso persistente también es un hallazgo común en estos pacientes. Pero, no se informaron previamente en un paciente con una mutación del gen MAP2K117. Además, la ausencia de vena cava inferior no se informó previamente en un paciente con síndrome de Noonan, no obstante, el síndrome de Noonan es la segunda causa no sindrómica más común de cardiopatía congénita 18. Esta última alteración podría ser un riesgo de trombosis venosa profunda idiopática 19, por lo que es necesario un seguimiento estricto del paciente.
A nivel oftalmológico, el paciente desarrolló blefaroptosis en la 19 semana de vida, que luego se reparó quirúrgicamente. Esta es una de las características externas más frecuentes en el síndrome de Noonan, por lo que se recomienda un examen oftalmológico temprano durante el diagnóstico del síndrome de Noonan 20. Similar a las características clínicas anteriores, esta característica no estaba relacionada previamente con la mutación del gen MAP2K1. En el sistema urinario, se presentó hidronefrosis prenatal, el cual está presente en aproximadamente en el 10% de los casos de síndrome de Noonan previamente reportados 21. También presentó criptorquidia unilateral, presente en 70% de los casos 21. Sin embargo, estas características tampoco están relacionadas con la mutación del gen MAP2K1.
Finalmente, en el sistema gastrointestinal, se presentó constipación y reflujo gastroesofágico con disfagia, que se iniciaron en los primeros meses de vida, similar a lo informado por Shah 22. Esta última presentación clínica podría estar relacionada con el rápido deterioro del crecimiento y el peso, como observamos en nuestro caso, por lo que una ingesta nutricional adecuada, al igual que en cualquier otro niño, es muy recomendable (23.
El gen MAP2K1 (OMIM 176872) transcribe una quinasa que fosforila las proteínas quinasas reguladas por señal extracelular 24. Se ha reportado casos raros de síndrome de Noonan negativo para PTPN11 y positivo para MAP2K1, y representan menos del 1% de la población de estudio 6,25,26. El fenotipo inicial de los pacientes con mutación MAP2K1 podría ser leve, pero durante el seguimiento presentarían más complicaciones 6, tal y como sucedió en nuestro caso. El síndrome de Noonan es una condición genéticamente heterogénea, es por ello que el enfoque de diagnóstico que utiliza paneles NGS es útil, rentable y rápido 27. Esta metodología ayudó en el diagnóstico final.
El gen CBL (OMIM 613563) es una ubiquitina ligasa E3 que regula negativamente la señalización intracelular contracorriente de los receptores tirosina quinasas, pero también contribuye al tráfico de señales 28. Las mutaciones heterocigotas de la línea germinal en CBL pueden ser la base de un fenotipo con características clínicas que se ajustan o que se superponen parcialmente al síndrome de Noonan (29, 30). Si bien, las mutaciones heterocigotas de la línea germinal en CBL están relacionadas con la leucemia mielomonocítica juvenil 31, que tiene una variante patógena conocida en el gen MAP2K1, la variante CBL de significancia desconocida detectada en el paciente, es probablemente un hallazgo que no afecta la salud. Sin embargo, podría existir un riesgo de leucemia en el futuro, particularmente con las mutaciones CBL. Se necesita un equipo multidisciplinario para el seguimiento del síndrome de Noonan, centrado en las posibles complicaciones conocidas, que incluye: neonatólogo, pediatras, genetistas, rehabilitadores, cardiólogos, neurólogos, gastroenterólogos y hemato-oncólogo.
El síndrome de Noonan es un trastorno genético común caracterizado por una gran variedad de hallazgos clínicos. El pronóstico varía dependiendo de las complicaciones que se presenten, el desarrollo de complicaciones sistémicas puede ser perjudicial si no se diagnostica a tiempo. El presente caso destaca la gran variedad de características fenotípicas que condicionaron el diagnóstico realizado al nacer. Además, algunas de las características clínicas de esta presentación moderada-severa podrían estar relacionadas con la mutación en el gen MAP2K1, que finalmente confirma el diagnóstico genético. La importancia de hacer un diagnóstico temprano radica en conocer la causa, guiar el manejo, prevenir complicaciones e informar el riesgo de recurrencia a los padres. Para hacer un diagnóstico temprano y una terapia rápida y adecuada, es fundamental que todos los pediatras conozcan esta enfermedad y su variedad clínica.

