










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Peruana de Ginecología y Obstetricia
versión On-line ISSN 2304-5132
Rev. peru. ginecol. obstet. vol.59 no.4 Lima oct./dic. 2013
CASO CLÍNICO
Tumor del saco vitelino puro con síndrome de Turner mosaico X0/XY
Pure yolk-sac tumor with XO/XY Turners syndrome mosaicism
Víctor Figueroa Zevallos1, Haydee Castro La Rosa1, Esperanza Torres Arones1
1 Servicio de Ginecología especializada, Hospital Nacional Edgardo Rebagliati Martins, EsSalud, Lima, Perú
Resumen
El síndrome de Turner es una afección genética por falta de un cromosoma X. Algunos casos se presentan con mosaicos XY, aumentando el riesgo de tumores gonadales de comportamiento maligno. Es importante determinar la presencia del cromosoma Y para realizar la extirpación profiláctica de las gónadas disgenésicas. Presentamos el caso de una paciente con síndrome de Turner, mosaico 45,X0/46,XY, asociado a tumor del saco vitelino puro gigante. Esta asociación es un caso extremadamente raro de tumor maligno en una paciente con disgenesia gonadal mixta.
Palabras clave: Síndrome de Turner, mosaicismo, disgenesia gonadal, tumor del saco vitelino.
Abstract
Turners syndrome is a genetic disorder caused by lack of an X chromosome. In some cases it is present with XY mosaicism, with increased risk of malignant gonadal tumors. It is important to determine the presence of the Y chromosome to perform prophylactic removal of the dysgenetic gonads. We present the case of a patient with Turners syndrome 45,X0/46,XY mosaicism associated to pure giant yolk sac tumor. This association of malignancy in a patient with mixed gonadal dysgenesia is extremely rare.
Keywords: Turners syndrome, mosaicism, gonadal dysgenesia gonadal, yolk sac tumor.
INTRODUCCIÓN
El síndrome de Turner (ST) fue descrito en 1938 por el endocrinólogo Henry Turner, pero recién en 1959, Ford, mediante un cariotipo, determinó que era debido a la presencia de un solo cromosoma X. Posteriormente se determinó, en un mismo individuo, la presencia de células con contenido genético y cromosómico diferente, estableciéndose la existencia de variedades o mosaicos. La variedad 45,X0/46,XY representa 5% de todos los casos de ST(1).
La disgenesia gonadal puede resultar en anomalías de la diferenciación sexual con expresión de diversas formas genotípicas y fenotípicas. En los casos sin ambigüedad sexual el diagnóstico se hace en la pubertad, cuando una de las principales manifestaciones es la amenorrea. Sin embargo, los problemas de crecimiento y el hallazgo de malformaciones menores o mayores podrían repercutir en un diagnóstico más temprano (2).
La presencia del cromosoma Y en pacientes con disgenesia gonadal aumenta el riesgo de tumores germinales, en especial de gonadoblastoma. Aunque este es un tumor de potencial de malignidad bajo, puede transformarse en disgerminoma invasivo en 60% de los casos y en otros tumores malignos de células germinales. El tumor del saco vitelino es una neoplasia de células germinales que ocurre generalmente en niñas y mujeres jóvenes, ya sea en combinación con otros tumores de células germinales o en su forma pura(3). Esta neoplasia, de presentación rara, agrupa neoplasias teratoides con diversos tipos de tejido endodérmico. Clínicamente son de extensión rápida, extremadamente malignas y con supervivencia a los 3 años de 13% (4,5).
Presentamos el caso de síndrome de Turner y tumor del saco vitelino cuya asociación es sumamente rara, habiendo encontrado solo 2 publicaciones de reporte de caso en los buscadores Pubmed y Lilacs.
REPORTE DE CASO
La historia clínica es de una paciente de 25 años con un tiempo de enfermedad de tres meses, con dolor pélvico, sensación de tumoración pélvica y dificultad para miccionar y defecar. Tenía como antecedente padecer de amenorrea primaria, sin mayor estudio previo y con diagnóstico de ovarios infantiles.
El examen físico mostró talla 1,40 m y peso 46,5 kg. Tenía implantación baja del cabello, cuello discretamente alado y pobre desarrollo mamario (Tanner 2-3). En el examen de abdomen fue evidente una tumoración abdominopélvica de 20 cm, que llegaba hasta la cicatriz umbilical, poco móvil y de consistencia dura. Los genitales externos femeninos tenían apariencia normal, la vagina era hipotrófica, con cérvix pequeño, tumoración dura poco móvil de 20x20 cm, que ocupaba toda la pelvis hasta la cicatriz umbilical.
Se realizó estudio del cariotipo, encontrándose 50% 45,X0 y 50% 46,XY, que correspondía a disgenesia gonadal mixta o ST mosaico. El estudio de ultrasonido transvaginal reveló una gran tumoración heterogénea multinodular de 14x8x13 cm con múltiples áreas hipoecoicas y zonas de calcificación hiperecoicas. El Doppler del vaso de la tumoración descrita tenía IP: 0,81, IR: 0,51 (figura 1). La tomografía abdominopélvica mostró lesión compleja que ascendía desde el piso pélvico hasta el nivel del mesogastrio, con áreas líquidas centrales, septos irregulares y paredes engrosadas; medía 120 mm de diámetro. No se delimitó contenido uterino u ovárico y había ausencia de lesión focal en el hígado, bazo, páncreas, suprarrenales y riñones. El riñón tenía configuración en herradura. No hacía adenomegalias ni líquido libre.
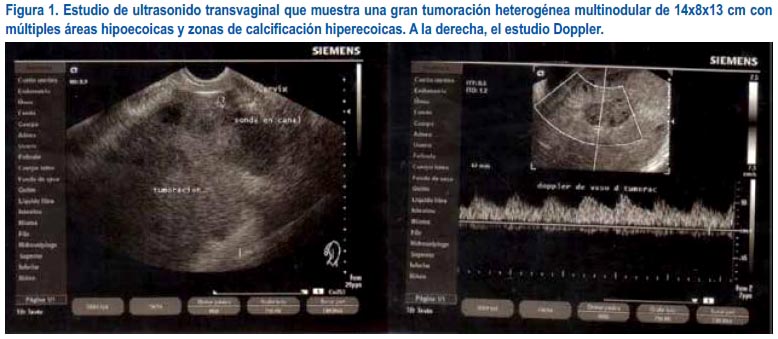
Los resultados de marcadores tumorales fueron los siguientes: βhCG (-), antígeno de cáncer (CA-125): 57 U/mL; alfa feto proteína (AFP): 211 ng/mL (VN: 0–10,9); lactato deshidrogenasa (LDH): 787 UI/L (VN: 100-210). En lo referente al perfil hormonal, se encontró: estriol libre <0,25 pg/mL; hormona tiroestimulante (TSH) 0,481 mU/L (VN: 0,4–4,0); hormona luteotrófica (LH)) 10 UI/L (VN: 1,1–11,6), hormona foliculoestimulante (FSH) 34 UI/L (VN: 2,8–11,3), prolactina (PRL) 7,35 ng/mL (VN: 4,5–33).
La paciente ingresó a sala de operaciones, realizándose citorreducción subóptima: lavado peritoneal + histerectomía total + salpinogooforectomía + omentectomía total + apendicetomía. Se encontró tumoración ovárica sólida friable, de aproximadamente 25x20x20 mm que ocupaba toda la cavidad pélvica, firmemente adherida a todas las estructuras adyacentes, las cuales incluían peritoneo parietal, epiplón, vejiga, útero, recto sigmoides, intestino delgado, paredes pélvicas laterales y fondo de saco de Douglas. Infiltraba y hacía cuerpo con el útero, el cual estaba comprometido por tumoración en sus 2/3 superiores. No se logró identificar ovario sano. Vejiga y recto sigmoides comprometidos a través de implantes en su superficie peritoneal, sin compromiso luminal. Dadas las características del componente adherencial, no se realizó la linfadenectomía pélvica. La paciente tuvo evolución favorable, saliendo de alta ocho días después de la intervención quirúrgica.
El examen macroscópico reveló un tumor lobulado de 20 cm, sólido, de consistencia blanda. En los cortes seriados se apreció una superficie heterogénea de color gris blanquecino con numerosas áreas hemorrágicas y necróticas, de aspecto microquístico (figura 2A). Histológicamente, la neoplasia estaba constituida por células atípicas, con vacuolas citoplasmáticas y núcleos pleomórficos, que adoptaban un patrón tubular perivascular, correspondiendo a tumor del saco vitelino (figura 2B). Dicha neoplasia se encontraba infiltrando útero y parte del epiplón. El apéndice cecal estaba libre de neoplasia. No se encontró gónada contralateral.
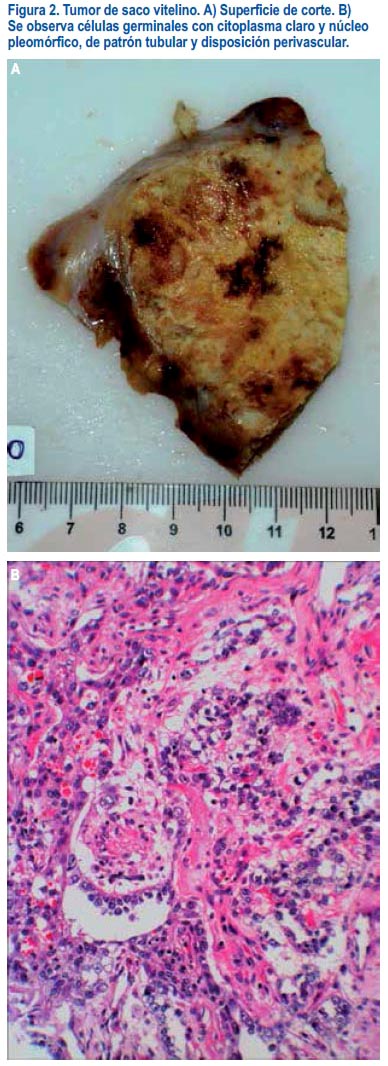
A la fecha, la paciente ha recibido tres cursos de quimioterapia, y aún continúa con valores de alfafetoproteína elevada y tumoración pélvica residual.
DISCUSIÓN
La disgenesia gonadal es un defecto embrionario en el desarrollo de las gónadas. Las fallas en el desarrollo gonadal pueden resultar en gónadas extremadamente hipoplásicas y disfuncionales o puede dar lugar a desarrollo testicular incompleto (testículos disgenéticos). Se llama disgenesia gonadal pura a los pacientes fenotípicamente femeninas con cariotipo 46,XX o 46,XY y gónadas en cordón bilaterales(6).
Se ha denominado disgenesia gonadal mixta a un grupo heterogéneo de anomalías gonadales, cromosómicas o fenotípicas, caracterizado por la presencia de testículo disgenético de un lado y una gónada en cordón o ausente del otro, persistencia de órganos derivados de los conductos de Müller y grados variables de ambigüedad sexual (7,12).
En general, la disgenesia gonadal mixta (DGM) se asocia a un cariotipo 45,X0/46,XY, con un espectro fenotípico que varía desde genitales externos femeninos a masculinos prácticamente normales, pasando por diferentes grados de ambigüedad genital, presencia de una gónada disgenética y un testículo disgenético contralateral o una banda de testículos disgenéticos bilateral, con elevado riesgo de desarrollar neoplasias germinales(8,12). La DGM debe ser considerada en el diagnóstico neonatal ante una ambigüedad sexual o puede sospecharse en la pubertad cuando hay pobre desarrollo de caracteres sexuales secundarios o amenorrea primaria(9).
El brazo corto del cromosoma Y contiene el gen para iniciar la diferenciación testicular conocida como región determinante del sexo del cromosoma Y (SRY). Una anomalía en el brazo corto del cromosoma Y daría lugar a la presencia de estrías gonadales o disgenesia testicular y los genitales externos e internos serían ambiguos, pues las secreciones del factor inhibidor de Müller y de testosterona estarían alterados por los testículos anormales. No se forman los testículos y por eso se desarrolla un fenotipo femenino. La pérdida parcial del cromosoma X en algunas células (X0/XY) producirían DGM, la que daría lugar a una de estas variantes: testículo inmaduro de un lado y gónada rudimentaria del otro; agenesia gonadal unilateral; gónadas hipoplásicas bilaterales intraabdominales con elementos rudimentarios testiculares en una de ellas o gonadoblastoma unilateral o bilateral(11). En el caso de nuestra paciente, no hemos podido establecer cuál fue la forma de presentación inicial, pero al parecer pudo haberse tratado de agenesia gonadal unilateral con gónada única disgenésica, ya que no se identificó otra gónada.
Las gónadas disgenésicas con algún fragmento del cromosoma Y conllevan un riesgo alto de tumores gonadales, principalmente de gonadoblastoma y disgerminoma. Se ha encontrado fragmentos del cromosoma Y en mosaicos 45,X0/46,XY usando técnicas de PCR y mediante pruebas específicas para búsqueda del cromosoma Y, con Southern blot e inmunofluorescencia con hibridización in situ(10,12). Ante el riesgo de desarrollar una neoplasia maligna tempranamente, se recomienda la gonadectomía profiláctica; sin embargo, un estudio de cohortes de 597 mujeres no encontró casos de gonadoblastoma o disgerminoma en 29 mujeres con presencia de cromosoma Y ni en las 568 mujeres restantes sin cromosoma Y(10).
Los tumores del saco vitelino representan neoplasias de crecimiento rápido, histológicamente diferentes y de presentación más rara que los gonadoblastomas. Los casos llamados puros, en el que el tejido tumoral es parecido al saco vitelino, son aún más raros, y la mayoría son de tipo mixto correspondiente a tejidos de diferenciación somática y mesénquima. Estas neoplasias característicamente presentan niveles elevados de alfafetoproteína, que es utilizado como marcador tumoral de seguimiento(5).
Se ha publicado sobre pacientes con disgenesia gonadal mixta y tumores germinales -como gonadoblastoma y disgerminoma- en países de América Latina, mas no en nuestro país(13). Representa más rara la asociación de síndrome de Turner con tumor del saco vitelino, habiéndose hallado solamente dos publicaciones de reporte de casos en el buscador de Medline y Lilacs de desarrollo de un tumor del saco vitelino puro en un paciente con síndrome de Turner mosaico con un fragmento de cromosoma Y(10,14), por lo que este caso representa la tercera publicación y la primera de nuestro país y América Latina.
REFERENCIAS BIBLIOGRÁFICAS
1. Firth H, Hurst J. Turner syndrome 45 X and variants. Oxford desk reference. Clinical genetics. Cap 5. First Edition. NY: Oxford University Press. 2007:558-60. [ Links ]
2. Soraes H, Maia A, Campos M, Doria S, Lopes JM, Fontoura M. Clinicopathological features of 45,X/46,Xidic(Y) mosaicism and therapeutic implications: case report. Sao Paulo Med J. 2008;126(5):297-9. [ Links ]
3. Rocco RM, Do Nacimento IT, Nunes MV, Piñero L, Durado A, Bianco B. Y chromosome in Turner Syndrome: review of the literature. Sao Paulo Med J. 2009;127(6):373-8. [ Links ]
4. Sarria J, Spitale L. Endodermal sinus tumor. Eur J Gynaecol Oncol. 1986;7(1):36-9. [ Links ]
5. Kurman RJ, Norris HJ. Endodermal sinus tumor of the ovary: a clinical and pathology analysis of 71 cases. Cancer. 1976;38:2404-19. [ Links ]
6. Federman D. Primary amenorrhea. Adolescent Gynecology and Endocrinology Part I. Western J Med. 1979;131(5):411-6. [ Links ]
7. Ribeiro de Andrade J, Guerra-Junior G, Trevas Masiel-Guerra A. 46,XY and 45,X/46,XY testicular dysgenesis: similar gonadal and genital phenotype, different prognosis. Arq Bras Endocrinol Metab. 2010;54(3):331-4. [ Links ]
8. Gelincik I, Ozen S, Bayram I. Left ovarian gonadoblastoma with yolk sac tumor in a young woman. Indian J Pathol Microbiol. 2010;53:345-6 [ Links ]
9. Nunez Lipay M, Bianco B, Verreschi I. Disgenesias Gonadais e Tumores: Aspectos genéticos e clínicos. Arq Bras Endocrinol Metab. 2005;49:60-70. [ Links ]
10. Ito K, Kawamata Y, Osada H, Ijichi M, Takano H, Sekiya S. Pure yolk sac tumor of the ovary with mosaic 45X/46X+mar Turners syndrome with a Y-chromosomal fragment. Arch Gynecol Obstet. 1998;262:87-90. [ Links ]
11. Gonzales P, Quesada M, Cabrera R, Bello D. Disgenesia gonadal mixta con formula cromosómica 45X/46X, (MAR). Presentación de una paciente. Rev Cubana Endocrinol. 2002;13(3):231-7 [ Links ]
12. Caglayan B, Demiryilmaz F, Kendirci M, Ozyazgan I, Akalin H, Bittmann S. Mixed gonadal dysgenesis with 45, X/46, X, IDIC (Y) karyotype. Genetic Counseling. 2009;20(2):173-9. [ Links ]
13. Vidaurri H, Martínez A, Sadowinski S. Gonadoblastoma y disgenesia gonadal mixta. Bol Med Hosp Infant Mex. 1997;54(9):439-42. [ Links ]
14. Ono T, Sakai N, Hayashi Y, Saito M, Kawagoe S, Hiroi M. 45X0/46Xdic(Yq) mosaicism in Turners phenotype with endodermal sinus tumor od the ovary. Gynecol Obstet Invest. 1989;27(1):45-7. [ Links ]
Artículo recibido el 24 de mayo de 2013 y aceptado para publicación el 20 de agosto de 2013.
Correspondencia:
Dr. Víctor Figueroa Zevallos
Celular: 999 046 349
Correo electrónico: vfigueroaz@hotmail.com
