










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Peruana de Ginecología y Obstetricia
versión On-line ISSN 2304-5132
Rev. peru. ginecol. obstet. vol.60 no.2 Lima abr. 2014
ARTÍCULO DE REVISIÓN
Desarrollo placentario temprano: aspectos fisiopatológicos
Early placenta development: pathophysiological aspects
John Apaza Valencia1
1 Ginecólogo Obstetra, Hospital III Honorio Delgado de Arequipa, Perú.
2 Universidad Nacional de San Agustín, Arequipa, Perú.
RESUMEN
El éxito de la gestación humana es el resultado de una sucesión de procesos que se dan en forma coordinada en tiempo y espacio en la interfase materno, placentario, embrionario-fetal. El fallo de esta sucesión de procesos determina una inadecuada placentación, preeclampsia y restricción del crecimiento intrauterino. En este artículo se revisa aspectos fisiopatológicos del desarrollo placentario temprano.
Palabras clave: Trofoblasto, desarrollo placentario, preeclampsia, restricción del crecimiento intrauterino.
ABSTRACT
Successful human pregnancy is the result of a series of processes that occur in a coordinated manner in time and space in the embryonic, placental, fetal-maternal interface. Failure of these series of processes determines inadequate placentation, preeclampsia and intrauterine growth restriction. Pathophysiological aspects of early placental development are reviewed in this article.
Keywords: Trophoblast, placental development, preeclampsia, intrauterine growth restriction.
INTRODUCCIÓN
La placenta es un órgano fundamental para el desarrollo embrionario y fetal, y el éxito de la gestación humana es el resultado de una sucesión de procesos que se dan en forma coordinada en tiempo y espacio en la interfase materno, placentario, embrionario, fetal. Este proceso empieza desde la adaptación temprana de preparación del endometrio para la implantación del blastocisto. El desarrollo placentario temprano crea un microambiente celular, molecular, bioquímico, reológico y otros que es susceptible a factores ambientales de su entorno celular, factores maternos internos y externos, factores fetales, inmunológicos, genéticos, epigenéticos, que a medida que transcurre la gestación pueden ocasionar significativos cambios fisiopatológicos que llevan a un mal resultado gestacional. Este conocimiento debe ser necesario para quienes tratamos gestantes de riesgo alto y bajo de morbimortalidad. Por lo tanto, comprender el desarrollo placentario temprano resulta fundamental para entender mecanismos fisiopatológicos de preeclampsia, restricción del crecimiento intrauterino (RCIU), pérdida gestacional temprana, entre otros. En esta revisión pretendemos analizar desde un punto de vista fisiopatológico el desarrollo placentario temprano.
DESARROLLO PREIMPLANTACIONAL
El desarrollo preimplantacional humano es el primer paso del desarrollo del individuo y está dado por los siguientes eventos importantes: maduración final del ovocito, fertilización, transición de ovocito a zigoto, proliferación, diferenciación celular y formación del blastocisto. En este último se originan las células trofoblásticas y tiene una importante contribución en los tejidos extraembrionarios (placenta y membranas) y procesos de desarrollo temprano de la gestación.
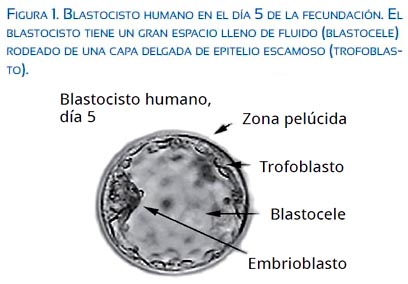
En la primera semana de formación del blastocisto humano (día 5 posfecundación), el trofoblasto está formado por una capa externa de células epiteliales escamosas aplanadas adyacentes una con otra y contenido dentro de la zona pelúcida (figura 1). Esta capa de células que se encuentran junto con el ectodermo del embrión forma lo que también se denomina trofoectodermo y será el epitelio responsable para la evolución de la placenta(1). Una condición previa y necesaria para la implantación es que se pierda la zona pelúcida y que el blastocisto se ponga en contacto directo con el endometrio.
DECIDUALIZACIÓN
La decidualización es una precondición para el establecimiento exitoso de la gestación y consiste en modificar las células estromales endometriales, glándulas uterinas, vasos y la población de células inmunes uterinas. En humanos, la decidualización es independiente de la presencia del blastocisto y empieza en la fase secretoria tardía del ciclo menstrual, la cual es regulada por progesterona y agentes regulatorios que elevan los niveles de cAMP.
Estos mecanismos moleculares locales aún son desconocidos. Sin embargo, algunos factores paracrinos/autocrinos han sido identificados en este proceso en el que participan citoquinas, interleuquina 11, activina A, factor β monoclonal supresor no específico (MNSFβ).
Las proteasas producidas localmente, como la proproteína convertasa 6 y metaloproteinasas de matriz (MMP1, MMP10 y MMP14), posibilitan la expansión del tejido y el procesamiento de moléculas reguladoras(2).
La decidualización está caracterizada por una disminución de linfocitos Th1 y un incremento de linfocitos Th2. Están presentes varios tipos de leucocitos, como linfocitos T, pocos linfocitos B, macrófagos y células asesinas naturales (NK). Sin embargo, después de la ovulación se ha observado un incremento de las células NK, las cuales se diferencian en células NK uterinas (uNK), que son fenotípicamente y funcionalmente diferentes de aquellas circulantes y no tienen actividad citolítica; están en contacto cercano con células estromales, trofoblasto, rodeando vasos y glándulas. Secretan múltiples citoquinas importantes en la angiogénesis. Su presencia en la fase secretoria tardía y decidual temprana parece ser importante en el mantenimiento de la decidualización. Contrariamente, la muerte de las células uNK resulta en un evento temprano del inicio de la menstruación(3). El incremento del número de células uNK ha sido asociado con falla reproductiva(4).
IMPLANTACIÓN DEL BLASTOCISTO
La implantación del blastocisto consiste en la interacción de las células del trofoblasto y el endometrio. Se da en un periodo corto de tiempo denominado ventana de receptividad entre el día 20 y 24 del ciclo menstrual. Fuera de este periodo, el endometrio está cubierto por una delgada capa de muscina (glicocálix) que evita la adhesión del blastocisto.
La adhesión del blastocisto al endometrio depende de la interacción entre moléculas de adhesión, tales como selectinas, integrinas y trofininas(5) . El control de la implantación del blastocisto es ejercido por las citoquinas y quimoquinas. El óxido nítrico favorece la implantación, modulando la liberación de prostaglandinas, esteroidegénesis ovárica, proliferación celular uterina, secreción glandular, flujo sanguíneo y factores de crecimiento(6). La presencia de un factor lítico en la cavidad endometrial es esencial para la disolución de la zona pelúcida. Parece ser que la plasmina producida del plasminógeno sea este factor uterino, debido a que la plasmina muestra in vitro efectos líticos sobre la zona pelúcida.
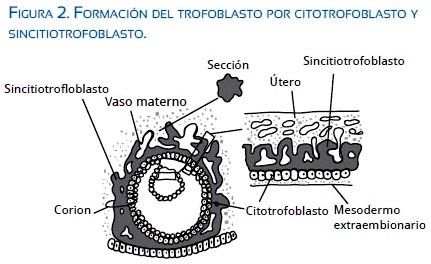
Una vez implantado el blastocisto, la capa de trofoblasto se diferencia en dos capas distintas: a) sincitiotrofoblasto (ST), que es la más externa; está formada por células multinucleares alineadas y hace contacto directo con la sangre materna; y, b) el citotrofoblasto (CT), que es la capa más interna compuesta por células mononucleares. Las células progenitoras de CT se fusionan unas con otras para formar el ST (figura 2). Se ha encontrado ST desorganizado, índice bajo de fusión, con pocos núcleos y disregulación de genes en la RCIU(7).
En la fase invasiva de implantación, el trofoblasto libera señales paracrinas que modulan la expresión génica celular del estroma decidual (CXCL1, GRO1 e IL8,CXCR4). Las células CT producen gonadotropina coriónica humana (hCG) y gonadotropina coriónica humana hiperglicosilada, promoviendo la formación de células del ST(8). La hCG y la hCG hiperglicosilada controlan el establecimiento de la placentación hemocorial eficientemente, controlan la formación de vellosidades placentarias y actúan como un agente inmunosupresivo(9). Paralelamente, el ST secreta enzimas proteolíticas que rompen la matriz extracelular; de esta manera permite primero, el pasaje del blastocisto a la pared endometrial en su totalidad, generando espacios lacunares que se llenan de sangre materna; en segundo lugar, permite la nutrición del embrión mediante la lisis de células epiteliales y deciduales adyacentes (nutrición histiotrofa); y, en tercer lugar, secreta lactógeno placentario y hCG que mantiene la decidua y la producción de progesterona por el cuerpo lúteo. Posteriormente, el trofoblasto fagocita células sanguíneas (nutrición hematotrofa)(10).
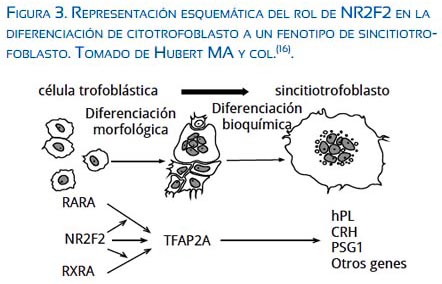
Alrededor del día 14 después de la implantación, el CT da lugar a un grupo celular denominado trofoblasto extravelloso (EVT). La diferenciación de CT a ST o EVT es controlado por diferentes agentes, tales como factores de transcripción, genes específicos, hormonas, factores de crecimiento citoquinas y niveles de oxígeno. Se ha encontrado in vitro que la diferenciación de CT a un fenotipo de ST está relacionado hasta en 8 veces con niveles elevados de mRNA del receptor hormonal nuclear NR2F2, alcanzando su pico máximo al primer día de la diferenciación, comparado al citotrofoblasto no diferenciado (figura 3)(11).
El ST ejerce un rol crucial en el intercambio feto-materno. Las células EVT tienen un fenotipo invasivo e infiltran el tejido uterino materno por dos vías: a) la primera por migración endovascular, formando columnas de células trofoblásticas que ocluyen las arterias espirales; b) segunda por invasión intersticial, la que requiere un proceso bioquímico de adhesión al citotrofoblasto y migración a través de la decidua y miometrio asociado a degradación proteolítica de componentes de la matriz extracelular(12) . La invasión del EVT es un proceso coordinado en tiempo y espacio y la alteración de este ajustado control está asociado con pérdida gestacional temprana y tardía, preeclampsia y RCIU (13-15). Los factores que controlan la invasión del EVT no son bien entendidos. Sin embargo, se ha descrito que las células EVT expresan en su superficie el HLA-C como molécula relevante y las células uNK expresan una variedad de receptores, incluyendo los receptores de la familia KIR (receptor de inmuno globulina asesina) que reconocen moléculas de HLA-C (figura 4) . De la interacción ligando-receptor se producen señales activadoras e inhibitorias que influencian el grado de invasión trofoblástica de células EVT en la pared de arterias espirales.
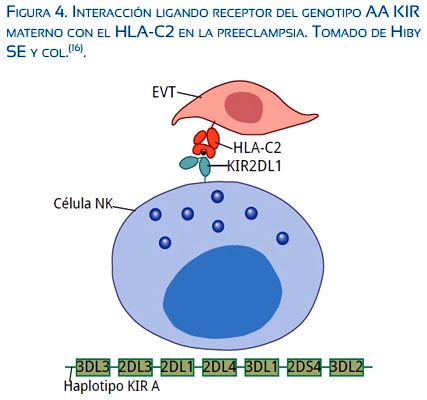
Así, la combinación del genotipo AA KIR materno con el HLA-C2 está significativamente asociado a preeclampsia. Lo cual da lugar a un defecto patológico primario, como es la falla de transformación de las arterias espirales uterinas(16).
El proceso de invasión de EVT evita el flujo sanguíneo materno y crea un medioambiente bajo en O2 hasta las 10 semanas de gestación; tal concentración de oxígeno puede ser tan baja y llegar a 1 o 2%. Se ha cuantificado el efecto de la edad gestacional y la concentración de oxígeno sobre el brote de EVT y se encontró en cultivos bajos de oxígeno una disminución en la frecuencia y tamaño del brote de EVT y una respuesta diferencial significativa a la concentración de oxígeno en placentas menores a 11 semanas de gestación. Sin embargo, no se observó respuesta diferencial en las vellosidades de placentas de 11 a 12 semanas. Por lo tanto, el oxígeno y la edad gestacional son un mecanismo de regulación del brote de EVT(17). La hipoxia actúa modificando la expresión génica, por ejemplo, estimula la expresión del factor 1 inducible por hipoxia (HIF-1), el cual mantiene la proliferación del CT y así junto con TGF-β3 previene la diferenciación. Sin embargo, este mecanismo de regulación por hipoxia no ocurre antes de las 7 semanas.
El medioambiente local rico en citoquinas y quimoquinas del endometrio decidualizado facilita la invasión trofoblástica y, en el miometrio, el EVT produce notables cambios de los vasos uterinos, estableciendo una circulación intervellosa periférica alrededor de las 9 semanas y se extiende por toda la placenta después de las 12 semanas (figura 5)(18,19). Entre las 8-10 semanas de gestación se encontró que el EVT y el citotrofoblasto fueron los mayores productores de factores de crecimiento angiogénicos, que probablemente contribuyen al desarrollo vascular placentario y remodelación de las arterias espirales mediante el reemplazo de células endoteliales de vasos uterinos y el músculo vascular por material amorfo(20,21). Se ha encontrado que este proceso es regulado por el factor de crecimiento endotelial vascular (VEGF)(22). La máxima actividad trofoblástica se da a nivel central del lecho placentario y posteriormente se extiende en forma centrífuga hacia la periferia. La infiltración trofoblástica del miometrio es progresiva y limitada hasta las 18 semanas de gestación.
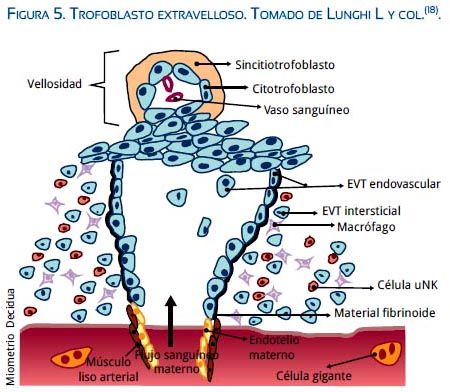
VELLOSIDADES CORIALES
Como resultado del proceso de implantación, proliferación y diferenciación del trofoblasto se forman las estructuras vellositarias. El trofoblasto del blastocisto implantado, junto con el mesodermo extraembrionario localizado internamente a este, forma lo que se denomina corion.
Cerca del final del segundo mes de gestación, las vellosidades cubren el corion completamente y son casi uniformes en tamaño, después del cual se comprimen y degeneran, formando el corion liso. Contrariamente, las vellosidades asociadas con la decidua basal proliferan formando el corion frondoso o placenta definitiva, al final del periodo embrionario (10 semanas posmenstruación). Se piensa que este desarrollo se deba a una diferencia del aporte nutricional después de la implantación.
La vellosidad primaria es al principio pequeña y avascular; aparece entre el día 12 y 15 pos-concepción y forma numerosas protrusiones digitiformes de solo trofoblasto que invaden la decidua uterina. Las vellosidades secundarias aparecen entre el día 16 y 21 de la gestación y está formado por columnas de CT, ST y mesodermo, incrementan en tamaño y dan ramificaciones. Se aperturan los vasos maternos para formar lagunas o espacios intervellosos. La sangre materna rodea las vellosidades y al mismo tiempo absorben de ella sustancias nutritivas para el desarrollo del embrión. Es el inicio de la circulación materno placentaria. La vellosidad terciaria aparece después del día 21 de la gestación, presenta ramificaciones y está formado por columnas de CT, ST, mesodermo e islotes vasculares que desarrollarán en vasos sanguíneos (figura 6). Alrededor del día 28 pos-concepción (6a semana posmenstrual), la sangre embrionaria es llevada del corazón primitivo a la vellosidad mediante ramificaciones de la arteria umbilical, plexo vascular de la vesícula vitelina hasta los capilares de la vellosidad, con retorno al embrión por la vena umbilical. Se establece la circulación feto placentaria. El espacio intervelloso es dividido internamente en compartimientos por los septos placentarios, pero estos no alcanzan la placa coriónica y hay comunicación entre los espacios intervellosos de diferentes compartimientos. A la vez, el espacio intervelloso es drenado por venas de la superficie de la decidua basal.
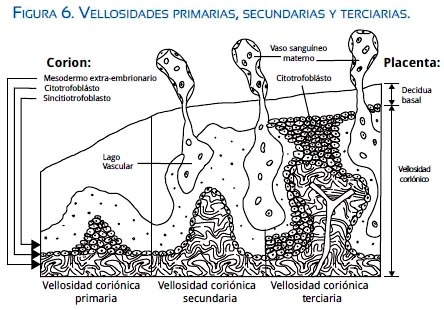
La primera generación de vellosidades terciarias se denominan vellosidades mesenquimales, cuya formación en adelante estará dada mediante lo que se denomina brote trofoblástico y se caracteriza por invasión mesenquimal y angiogénesis fetal local. Conforme avanza la gestación, la vellosidad mesenquimal dará lugar a la formación del árbol velloso(23).
La vellosidad mesenquimal es la primera estructura morfológica para un efectivo intercambio materno-fetal de nutrientes, gases y sustancia residuales. Esta primera generación de vellosidades mesenquimales se forman entre el día 20 y 42 posconcepción y revela la presencia de vasculogénesis (formación de novo de capilares de células precursoras mesenquimales), como un mecanismo subyacente de formación vascular.
Cerca de la 5a semana posconcepción (7 semanas posmenstrual), todas la vellosidades placentarias pertenecen al tipo mesenquimal, que en adelante pueden diferenciar a tipos especializados de vellosidades, como la vellosidad intermedia inmadura y de estas a vellosidad tronco.
Después del 4º mes de gestación, las vellosidades tienen forma de árbol, formado por una doble capa de ST y CT o células de Langerhans.
Las vellosidades pueden ser clasificadas por su relación en vellosidades flotantes, por encontrarse libremente en el espacio intervelloso, y vellosidades tronco de anclaje, cuya función es estabilizar la integridad anatómica de la interfase materno-placentaria (figura 7). La relación vellosidad flotante/vellosidad de anclaje incrementa según progresa la gestación, particularmente en el segundo y tercer trimestre(24).
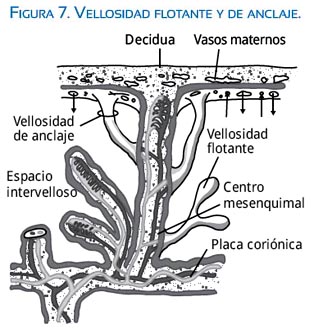
Los septos placentarios dividen la parte fetal de la placenta en 10 a 38 áreas convexas compuestas de lóbulos que darán lugar a los cotiledones. Cada cotiledón está formado de 2 o más vellosidades tronco principales y múltiples ramificaciones. Paralelamente, la parte vascular tiene similar desarrollo; la red capilar en todas las ramificaciones vellositarias se originan del tronco vascular de la vellosidad.
Empezando las 23 semanas posconcepción, el desarrollo de las vellosidades cambia de vellosidades mesenquimales a vellosidades intermedias maduras y de estas brotan las vellosidades terminales. Estas vellosidades son estructuras muy capilarizadas, de 100 μm de longitud y 80 μm de diámetro aproximadamente, con lo cual forman vellosidades altamente efectivas para el intercambio difusional materno-fetal (figura 8). El control del cambio de una vellosidad a otra aún sigue siendo un misterio(23).
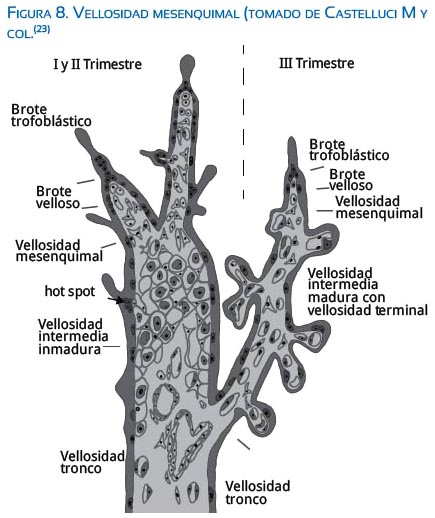
El cambio anormal retrasado o precoz hacia vellosidades intermedias maduras determina un mal desarrollo vellositario y está relacionado a RCIU, pérdida gestacional y pobre resultado neonatal(25).
La diferenciación de las vellosidades coincide temporalmente con el desarrollo de la arquitectura lobular y los procesos son probablemente entrelazados. Los lóbulos pueden ser identificados al inicio del segundo trimestre seguido del inicio de la circulación materna, cuya fuerza hemodinámica puede formar el árbol velloso. Hay evidencia que la sangre materna fluye por la parte central del lóbulo y se dispersa periféricamente con gradientes de concentración de oxígeno altos en el centro del lóbulo.
La constitución aproximada de la placenta a término es de 40 a 50% de vellosidades terminales, 25% de vellosidades intermedias maduras, 20 a 25% de vellosidades tronco, 5 a 10% de vellosidades intermedias inmaduras y menos del 1% de vellosidades mesenquimales. El área de superficie vellositaria es de 10 a 14 m2 y ello puede ser significativamente disminuido en casos de RCIU. Aquí refleja una reducción global del volumen placentario, volumen reducido del espacio intervelloso y puede mostrar también pobre crecimiento de vellosidades periféricas afectando los capilares, estroma y trofoblasto. En casos de preeclampsia, el área de superficie vellosa es normal(26) . El volumen placentario medido por ecografía a las 12 semanas es reducido en la RCIU, pero tiene un desarrollo similar al normal. El desarrollo placentario en preeclampsia es significativamente más grande a las 12 semanas y crece normal hasta las 16 semanas, deteniendo su crecimiento entre las 16 y 22 semanas. En los casos con RCIU y preeclampsia, el volumen placentario a las 12 semanas es reducido significativamente y el crecimiento hasta las 22 semanas es heterogéneo. Estos hallazgos sugieren que la preeclampsia y la RCIU se originan en el primer trimestre(37).
TROFOBLASTO
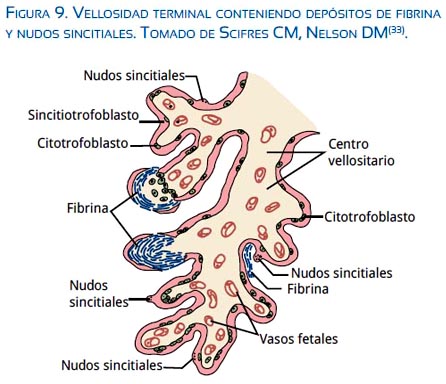
La integridad anatómica del sincitiotrofoblasto es fundamental para regular el intercambio materno fetal. El funcionamiento del trofoblasto como unidad estructural depende del posicionamiento estratégico de una variedad de transportadores, receptores y enzimas que se encuentran sobre la membrana plasmática de la microvellosidad, tanto del lado que baña la sangre materna como del lado de la membrana plasmática basal del centro de la vellosidad o ambos. La naturaleza endotelial de la superficie del ST permite la secreción direccional de hormonas, factores de crecimiento y citoquinas hacia la sangre materna e influencian el suministro de sustratos y crecimiento fetal(38). Las lesiones de las vellosidades pueden no ser acompañadas de un incremento compensatorio en la proliferación del CT y diferenciación en ST; esto predispone a una disminución del ST. La apoptosis de las vellosidades incrementa conforme progresa la gestación y sería parte del recambio normal del mismo(29). A partir del tercer trimestre, en algunas vellosidades el núcleo del ST tiene forma de racimo y son denominados agregados nucleares sincitiales (SNAs), uno de los cuales son los nudos sincitiales (figura 9). Esta formación protruye de la superficie del sincitiotrofoblasto al espacio intervelloso y constituye un signo morfológico característico de apoptosis (30). La presencia de nudos sincitiales tiene una correlación positiva con el tiempo y severidad de la hipertensión de la gestación, así como la presencia de fibrina en las vellosidades(31). La exposición a hipoxia e hiperoxia o especies reactivas de oxigeno induce la formación de nudos sincitiales, lo que apoya como mecanismo patogénico en la preeclampsia y la RCIU (32,33).
Las glicoproteínas específicas de la gestación (PSG) son miembros de los antígenos carcinoem-brionarios familia de genes semejante a inmunoglobulinas; comprende 11 genes en humanos; los PSG son producidos por el ST. Su importancia radica en que potencian o inhiben la respuesta inmune, regulan la función linfocitaria y evitan el ataque inmune materno. Niveles sistémicos bajos de PSG-β1 en humanos se relacionan con hipoxia fetal, RCIU, preeclampsia y aborto espontáneo(34,35). La proteína placentaria 13 (PP13) es otra proteína específica secretada por la microvellosidad del ST, que puede tener rol funcional inmunobiológico en el espacio intervelloso materno-fetal o desarrollo de la placenta. Niveles séricos maternos bajos de PP13 en el primer trimestre de la gestación están asociados con preeclampsia severa de inicio temprano o tardío. En casos de mutación de esta proteína, la frecuencia de preeclampsia es alta (36).
CONCLUSIONES
Las evidencias disponibles nos permiten comprender que las consecuencias fisiopatológicas del desarrollo placentario temprano dependen de un complejo proceso de formación de células del trofoblasto, de una adecuada decidualización del endometrio materno, implantación e invasión trofoblástica. Es indispensable una coordinada sucesión de eventos celulares para una adecuada remodelación de vasos espirales y establecer la circulación uteroplacentaria. Los mecanismos fisiopatológicos para una variedad de factores etiológicos que aún siguen en estudio son múltiples y la consecuencia anatómica en el desarrollo placentario probablemente sea común de lesión de la vellosidad y el sincitiotro-foblasto y, en el lado materno, un desarrollo vascular uterino anormal y estrés oxidativo.
REFERENCIAS BIBLIOGRÁFICAS
1. Zhang P, Zucchelli M, Bruce S, Hambiliki F, Stavreus-Evers A, Levkov L, Skottman H, Kerkelä E, Kere J, Hovatta O. Transcriptome profiling of human pre-im-plantation development. PLoS ONE. 2009;4(11);e7844.
2. Salamonsen LA, Dimitriadis E, Jones RL, Nie G. Complex regulation of decidualization: a role for cytokines and proteases – a review. Placenta. 2003;24(Suppl A):76-85.
3. King A. Uterine leukocytes and decidualization. Hum Reprod Update. 2000;6:28-36.
4. Quenby S, Farquharson R. Uterine natural killer cells, implantation failure and recurrent miscarriage. Reprod Biomed Online. 2006;13:24-8.
5. Aplin JD, Kimber SJ. Trophoblast-uterine interactions at implantation. Reprod Biol Endocrinol. 2004;2:48-59.
6. Khorram O. Nitric oxide and its role in blastocyst implantation. Rev Endocr Metab Disord. 2002;3:145-9.
7. Ruebner M, Strissel PL, Langbein M, Fahlbusch F, Wachter DL, Faschingbauer F, Beckmann MW, Strick R. Impaired cell fusion and differentiation in placentae from patients with intrauterine growth restriction correlate with reduced levels of HERV envelope genes. J Mol Med. 2010;88(11);1143-56.
8. Cole LA. Biological functions of hCG and hCG-related molecules. Reprod Biol Endocrinol. 2010;8:102. doi: 10.1186/1477-7827-8-102.
9. Cole LA. hCG, the centerpiece of life and death. Int J Endocrinol Metab. 2011;9(2):335-52.
10. Fernandez N, Cooper J. A critical review of the role of the major histocompatibility complex in fertilization, preimplantation development and feto – maternal interactions. Reprod Hum Update. 1999;3:234-48.
11. Hubert MA, Sherritt SL, Bachurski CJ, Handwerger S. Involvement of transcription factor NR2F2 in human trophoblast differentiation. PLoS ONE. 2010;5(2);e9417.
12. Lash GE, Otun HA, Innes BA, Bulmer JN, Searle RF, Robson SC. Inhibition of trophoblast cell invasion by TGFB1, 2, and 3 is associated with a decrease in active proteases. Biol Reprod. 2005;73:374–81.
13. Khong TY, Liddell HS, Robertson WB. Defective haemochorial placentation as a cause of miscarriage: a preliminary study. Br J Obstet Gynaecol. 1987;94:649–55.
14. Pijnenborg R, Anthony J, Davey DA, Rees A, Tiltman A, Vercruysse L, van Assche A. Placental bed spiral arteries in the hypertensive disorders of pregnancy. Br J Obstet Gynaecol. 1991;98:648–55.
15. Khong TY, De Wolf F, Robertson WB, Brosens I. Inadequate maternal vascular response to placentation in pregnancies complicated by preeclampsia and by small for gestational age infants. Br J Obstet Gynaecol. 1986;93:1049–59.
16. Hiby SE, Walker JJ, Oshaughnessy KM, Redman CW, Carrington M, Trowsdale J, Moffett A. Combinations of maternal KIR and HLA-C genes influence the risk of preeclampsia and reproductive success. JEM. 2004;200(8):957-65.
17. James JL, Stone PR, Chamley LW. The effects of oxygen concentration and gestational age on extravillous trophoblast outgrowth in a human first trimester villous explant model. Hum Reprod. 2006;21(10):2699-705.
18. Lunghi L, Ferretti ME, Medici S, Biondi C, Vesce F. Control of human trophoblast function. Reprod Biol Endocrinol. 2007;5:6.
19. Hess AP, Hamilton AE, Talbi S, Dosiou C, Nyegaard M, Nayak N, Genbecev-Krtolica O, et al Decidual stromal cell response to paracrine signals from the trophoblast: amplification of immune and angiogenic modulators. Biol Reprod. 2007 Jan;76(1):102-17.
20. Lash GE, Naruse K, Innes BA, Robson SC, Searle RF, Bulmer JN. Secretion of angiogenic growth factors by villous cytotrophoblast and extravillous trophoblast in early human pregnancy. Placenta. 2010;31(6);545-8.
21. Vicovac L, Aplin JD. Epithelial-mesenchymal transition during trophoblast differentiation. Acta Anat. 1996;156:202-16.
22. Fukushima K, Miyamoto S, Tsukimori K, Kobayashi H, Seki H, Takeda S, Kensuke E, Ohtani K, Shibuya M, Nakano H. Tumor necrosis factor and vascular endothelial growth factor induce endothelial integrin repertories, regulating endovascular differentiation and apoptosis in a human extravillous trophoblast cell line. Biol Reprod. 2005;73:172-9.
23. Castellucci M, Kosanke G, Verdenelli F, Huppertz B, Kaufmann P. Villous sprouting: fundamental mechanisms of human placetal development. Hum Reprod Update. 2000;6(5):485-94.
24. Joanna L James, P R Stone, L W Chamley. The effects of oxygen concentration and gestational age on extravillous trophoblast outgrowth in a human first trimester villous explant model. Hum Reprod. 2006;21(10);2699-705.
25. Kingdom JC, Kaufmann P. Oxigen and placental villous development: origins of fetal hipoxia. Placenta. 1997;18:613-26.
26. Mayhew TM, Ohadike C, Baker PN, Crocker IP, Mitchell C, Ong SS. Stereological investigation of placental morphology in pregnancies complicated by preeclampsia with and without intrauterine growth restriction. Placenta. 2003;24:219-26.
27. Hafner E, Metzenbauer M, Höfinger D, Munkel M, Gassner R, Schuchter K, et al. Placental growth from the first to the second trimester of pregnancy in SGA-foetuses and pre-eclamptic pregnancies compared to normal foetuses. Placenta. 2003;24(4):336–42.
28. Jones HN, Powell TL, Jansson T. Regulation of placental nutrient transport – a review. Placenta. 2007 Aug-Sep;28(8-9):763-74.
29. Mayhew TM, Leach L, McGee R, Ismail WW, Mykle-bust R, Lammiman MJ. Proliferation, differentiation and apoptosis in villous trophoblast at 13-41 weeks of gestation (including observations on annulate lamellae and nuclear pore complexes. Placenta. 1999 Jul-Aug;20(5-6):407-22.
30. Coleman SJ, Gerza L, Jones CJ, Sibley CP, Aplin JD, Heazell AE. Syncytial nuclear aggregates in normal placenta show increased nuclear condensation, but apoptosis and cytoskeletal redistribution are uncommon. Placenta. 2013 May; 34(5):449–55.
31. Correa RR, Gilio DB, Cavellani CL, Paschoini MC, Oliveira FA, Peres LC, et al. Placental morphometrical and histopathology changes in the different clinical presentations of hypertensive syndromes in pregnancy. Arch Gynecol Obstet. 2008 Mar;277(3):201-6.
32. Heazell AE. Formation of syncytial knots is increased by hyperoxia, hypoxia and reactive oxygen species. Placenta. 2007 Apr;28 Suppl A:S33-40.
33. Scifres CM, Nelson DM. Intrauterine growth restriction, human placental development and trophoblast cell death. J Physiol. 2009;587(14):3453–8.
34. Grudzinskas JG, Gordon YB, Menabawey M, Lee JN, Wadsworth J, Chard T. Identification of high-risk pregnancy by the routine measurement of pregnan-cy-specific beta-1-glycoprotein. Am J Ostet Gynecol. 1983;147:10-2.
35. Pihl K, Larsen T, Laursen I, Krebs L, Christiansen M.First trimester maternal serum pregnancy-specific be-ta-1-glycoprotein (SP1) as a marker of adverse preg-nancy outcome. Prenat Diag. 2009;29:1256-61.
36. Huppertz B, Meiri H, Gizurarson S, Osol G, Sammar M. Placental protein 13 (PP13): a new biological target shifting individualized risk assessment to personalized drug design combating pre-eclampsia. Hum Reprod Update. 2013;19(4):391-405.
Declaración: Se declara que el material contenido en el manuscrito no ha sido publicado previamente o remitido a otra revista biomédica.
Financiamiento: Autofinanciado.
Conflicto de intereses: ninguno.
Artículo recibido el 20 de enero de 2014 y aceptado para publicación el 1 de mayo de 2014.
Correspondencia:
Dr. John Apaza Valencia
Dirección: Urb. Quinta El Sol Mz B Lote 11, Cerro Colorado, Arequipa
john_apaza@hotmail.com

