










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Peruana de Ginecología y Obstetricia
On-line version ISSN 2304-5132
Rev. peru. ginecol. obstet. vol.60 no.4 Lima Oct./Dec. 2014
SIMPOSIO PREECLAMPSIA, VIEJO PROBLEMA AÚN NO RESUELTO: CONCEPTOS ACTUALES
Actualización en la fisiopatología de la preeclampsia
Pathophysiology of preeclampsia: update
Luis Martín Gómez Carbajal 1,2
1 Médico Asistente, División de Medicina Materno-Fetal, Departamento de Gíneco-Obstetricia, University of Tennessee Health Science Center, Memphis, Tennessee, EE UU.
2 Director de las Clínicas de Alto Riesgo, University of Tennessee Health Science Center, Memphis, Tennessee, EE UU.
RESUMEN
La preeclampsia constituye una de las complicaciones más frecuentes y a la vez más serias de la gestación y contribuye de manera significativa a la mortalidad materna y perinatal. No obstante los avances en el estudio de la preeclampsia, aún no está del todo esclarecido su mecanismo fisiopatológico. En este capítulo, intentamos revisar nuevas teorías propuestas acerca de su fisiopatología. Los aspectos genéticos y angiogénicos serán revisados en otros capítulos de este simposio.
Palabras clave: isquemia placentaria, disfunción endotelial, óxido nítrico, estrés oxidativo, estrés del retículo endoplásmico, Chlamydia pneumoniae, fetuína-A.
ABSTRACT
Preeclampsia is one of the most frequent and serious disorders of pregnancy. It is a significant contributor of maternal and perinatal mortality worldwide. An important amount of research has been devoted in the research of preeclampsia in the recent years; nonetheless, its pathophysiology is yet to be completely understood. In this review, we will discuss new proposed theories on the pathophysiology of preeclampsia. Genetic and angiogenic aspects of preeclampsia will be reviewed elsewhere in this issue.
Keywords: placental ischemia, endothelial dysfunction, nitric oxide, oxidative stress, endoplasmic reticulum stress, Chlamydia pneumoniae, fetuin-A.
INTRODUCCIÓN
La preeclampsia, definida como hipertensión arterial que usualmente debuta (o agrava la hipertensión pre-gestacional) a las ≥20 semanas de embarazo, es un síndrome inducido por la gestación.
A pesar de avances en la fisiopatología y manejo, la preeclampsia sigue afectando hasta 7% de todos los embarazos, y es una de las principales causas de mortalidad materna y perinatal en países en desarrollo y desarrollados(1-8).
Factores de riesgo
En la tabla 1 se señala los factores de riesgo relacionados a la preeclampsia. La probabilidad de preeclampsia con manifestaciones severas aumenta de manera sustancial en mujeres con historia de preeclampsia, diabetes mellitus, insuficiencia renal crónica, síndrome de anticuerpos anti fosfolípidos, obesidad, hipertensión crónica o embarazo múltiple.
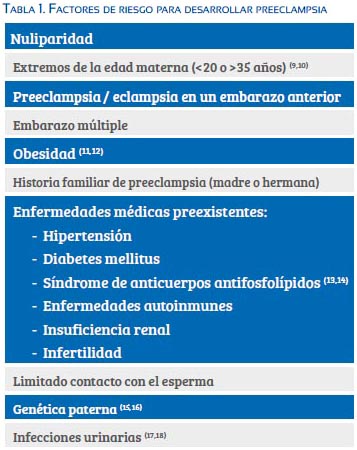
La preeclampsia usualmente es más frecuente en primigrávidas. Es probable que por un mecanismo inmune, la futura madre aprenda a tolerar los antígenos paternos presentes en el líquido seminal; la exposición limitada al esperma contribuiría como factor de riesgo para que la paciente desarrolle preeclampsia (tabla 1)(19). Ello explicaría por qué las mujeres con exposición limitada al esperma (primer coito y embarazo, embarazo tras inseminación artificial, multíparas que cambian de pareja) presenten mayor riesgo de preeclampsia(19).
Los varones que han engendrado un embarazo complicado con preeclampsia constituyen factores de riesgo para que una nueva pareja desarrolle preeclampsia en una futura gestación (genética paterna)(15).
La preeclampsia se puede presentar en grupos familiares, lo cual sugiere un componente genético(20). En estudios en gemelos, se estima que en 22% a 47% se puede heredar la preeclampsia(21). Estudios previos han demostrado asociaciones significativas entre preeclampsia y variantes del ADN en la cadena alfa 1 del colágeno (COL1A1), interleuquina-1 alfa (IL1A)(22), mutación del factor V Leiden, mutaciones de la sintetasa del óxido nítrico endotelial, antígeno leucocitario humano y de la enzima convertidora de angiotensina(23).
Avances significativos en la comprensión de la preeclampsia
A continuación describiremos una actualización de los mecanismos recientemente asociados con la preeclampsia. Los aspectos angiogénicos y genéticos serán abordados en otros segmento de este simposio.
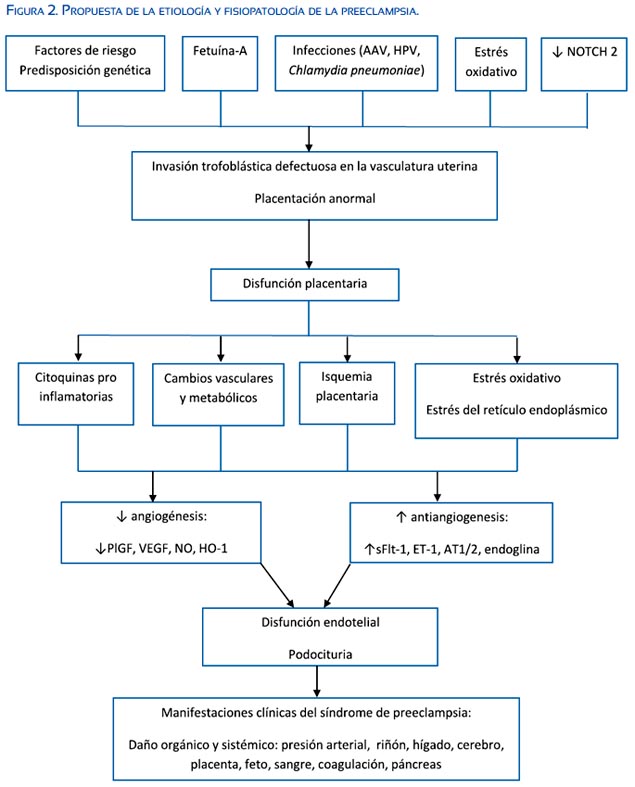
Se postula que la preeclampsia, sobre todo la de inicio temprano en el embarazo, se desarrolla en dos estadios(4,24-28). El primer estadio (antes de las 20 semanas) involucra una pobre invasión placentaria en el miometrio y la vasculatura uterina; es este estadio no hay manifestaciones clínicas.
El segundo estadio se manifiesta por las consecuencias de la pobre placentación, provocado por la relativa hipoxia placentaria y la hipoxia de reperfusión, lo cual resulta en daño al sincitiotrofoblasto y restricción del crecimiento fetal. El eslabón entre la hipoxia placentaria relativa y el síndrome clínico materno incluye una cascada de mecanismos secundarios incluyendo el desbalance entre factores pro-angiogénicos y anti-angiogénicos, estrés oxidativo materno, y disfunción endotelial e inmunológica(4,27).
Implantación anormal y vasculogénesis
Una de los mecanismos principales en la patogenia de la preeclampsia es el de la insuficiencia placentaria debida a una remodelación deficiente de la vasculatura materna de perfusión en el espacio intervelloso. En un embarazo normal, el citotrofoblasto fetal invade las arterias uterinas espirales maternas reemplazando el endotelio, y las células se diferencian en citotrofoblastos endotelioides(29). Este proceso complejo resulta en la transformación de vasos sanguíneos de pequeño diámetro y alta resistencia vascular en vasos de baja resistencia y alta capacitancia, asegurando así una distribución adecuada de la sangre materna a la unidad útero-placentaria en desarrollo. En la paciente predestinada a desarrollar preeclampsia, defectos en este proceso de transformación vascular aún no del todo comprendidos conducen a una entrega inadecuada de sangre a la unidad útero-placentaria en desarrollo e incrementa el grado de hipoxemia y estrés oxidativo y del retículo endoplásmico.
Los mecanismos exactos responsables de la invasión trofoblástica y remodelación vascular defectuosas no están del todo claros; sin embargo, recientes investigaciones permiten entender mejor los mecanismos anteriormente mencionados(4,24-28). Recientemente, investigadores han mostrado evidencia que la señalización NOTCH (NOTCH es una proteína transmembrana que sirve como receptor de señales extracelulares y que participa en varias rutas de señalización con el cometido principal de controlar los destinos de la célula) es vital en el proceso de invasión del trofoblasto y remodelación vascular. La ausencia de NOTCH2 se asociaría con reducción del diámetro vascular y afectaría la perfusión placentaria. Además, los investigadores demostraron en modelos de preeclampsia que los citotrofoblastos endovasculares y perivasculares carecían de JAG1 (que es un ligando del NOTCH2)(30).
Otros estudios sugieren que la variabilidad en los genes del sistema inmune que codifican las moléculas del complejo de histocompatibilidad y de los receptores de las células asesinas naturales puede afectar la placentación(31). Así, ciertos tipos de combinaciones entre moléculas del complejo de histocompatibilidad y genes de receptores de las células asesinas naturales se correlacionan con el riesgo de desarrollar preeclampsia, aborto recurrente y restricción del crecimiento fetal.
Activación y disfunción endotelial
El endotelio vascular materno en la paciente predestinada a desarrollar preeclampsia es objeto de variados factores que se generan como consecuencia de hipoxia e isquemia placentaria(4,27,32). El endotelio vascular tiene roles importantes, incluyendo el control del tono de la capa de músculo liso a través de la liberación de factores vasoconstrictivos y vasodilatadores, así como la liberación de diferentes factores solubles que regulan la anticoagulación, y funciones antiplaquetarias y fibrinolíticas. Se ha encontrado alteraciones de la concentración en la circulación de muchos marcadores de disfunción endotelial en mujeres que desarrollan preeclampsia(4,27,32). Esto sugiere que la preeclampsia es un desorden de la célula endotelial. El hecho que esta disfunción endotelial se pueda demostrar antes que la preeclampsia se desarrolle floridamente apoya esta teoría.
El estado materno influye la respuesta endotelial a factores derivados de la isquemia e hipoxia placentaria en la preeclampsia. Hay evidencia de que la obesidad incrementa el riesgo de preeclampsia. Un índice de masa corporal mayor de 39 incrementa en 3 veces el riesgo de preeclampsia(33). Los mecanismos que explican la influencia de la obesidad en la preeclampsia no están del todo elucidados.
Existe la teoría del rol protector de la endotelina tipo A (ETA), antagonista de la endotelina-1 (poderoso vasoconstrictor), en la preeclampsia. Una variedad de factores angiogénicos (sFlt-1, AT1AA, TNF-α) antagonizan la acción de ETA(34,35). En otros capítulos en esta edición se detalla con amplitud la influencia de factores angiogénicos en la fisiopatología de la preeclampsia.
Oxido nítrico
El óxido nítrico (NO) es un regulador importante de la presión arterial. La producción de NO está incrementada en el embarazo normal y probablemente relacionada a la vasodilatación fisiológica del embarazo. Se ha postulado que la deficiencia de NO predispondría la ocurrencia de preeclampsia. La inhibición crónica de NO sintetasa en ratas preñadas produce hipertensión asociada con vasoconstricción renal y periférica, proteinuria, restricción del crecimiento intrauterino y morbilidad fetal, de manera similar a los hallazgos presentes en preeclampsia(27). Sin embargo, es todavía controversial el concepto de reducción en la producción de NO en la preeclampsia; esto es debido a la dificultad en medir la actividad y producción de NO en la práctica clínica.
Estrés oxidativo y estrés del retículo endoplásmico
Se ha encontrado una concentración incrementada de muchos marcadores de estrés oxidativo en la preeclampsia, como los peroxinitritos(36,37). La concentración de peroxinitritos en el endotelio vascular es mucho más elevada en mujeres con preeclampsia que en aquellas con embarazos normales, lo cual coincide con concentraciones disminuidas de superóxido dismutasa y NO sintetasa(38). También hay evidencia de estrés oxidativo incrementado en modelos de hipertensión en roedores, lo que sugiere un lazo entre la hipoxia e isquemia placentaria, con la producción de sustancias oxígeno reactivas(27). El uso de antioxidantes como vitamina C y vitamina E no tiene efecto beneficioso en la prevención o tratamiento de preeclampsia(39). Sin embargo, el uso de tempol (mimético de la superóxido dismutasa) atenúa la respuesta hipertensiva del estrés oxidativo. De la misma manera, el uso de apocinina (inhibidor la nicotinamida adenina dinucleótido fosfato oxidasa) también atenúa la hipertensión arterial en modelos roedores(27). Está por determinar si la producción de sustancias oxígeno reactivas es la causa primaria o secundaria de la preeclampsia.
También, parece haber un exceso del estrés del retículo endoplásmico en mujeres que desarrollan preeclampsia en el embarazo temprano(40). El estrés del retículo endoplásmico activa un número de señales que buscan restaurar la homeostasis. Se ha propuesto que este mecanismo homeostático falla y que se activan vías apoptóticas que alteran la función placentaria en mujeres que desarrollan preeclampsia(40). Además, concentraciones bajas de manera crónica del estrés del retículo endoplásmico durante el segundo y tercer trimestres pueden derivan en restricción del crecimiento asociado a preeclampsia. Por otro lado, altas concentraciones de estrés del retículo endoplásmico conduce a la activación de vías proinflamatorias que pueden contribuir a la activación del endotelio materno.
Hemoxigenasa
Se ha postulado que el gen de respuesta de estrés, hemoxigenasa-1 (HO-1) y su producto catalítico, monóxido de carbono, estarían involucrados como factores protectores en la patogénesis de la preeclampsia(41). El bloqueo genético o farmacológico de HO-1 en modelos animales induce manifestaciones clínicas parecidas a la preeclampsia(41). HO-1 y sus derivados catalíticos brindarían protección contra la progresión hacia la preeclampsia al interferir en los mecanismos en que la hipoxia placentaria induce hipertensión(42-44). Así, de manera curiosa se sabe que la combustión de productos del tabaco, como el monóxido de carbono, reduce el riesgo de preeclampsia en más de 35%(43). Además, el daño celular en la vellosidad placentaria inducido por el factor de necrosis tumoral alfa (TNF-α) puede prevenirse al incrementar la expresión de la actividad de HO1(42). Las vías de la hemoxigenasa también inhiben la liberación de la forma soluble fms de tirosina quinasa-1 (sFlt-1) en modelos in vitro(44). La inducción de HO-1 o la administración crónica de metabolitos de la HO-1 mejoran la hipertensión en variados modelos animales de hipertensión debida a factores regulatorios similares a los observados en mujeres con preeclampsia. La administración crónica de inductores de HO-1 (como el cloruro de protoporfirina de cobalto IX) o de moléculas liberadores de monóxido de carbono atenúa significativamente la hipertensión inducida por isquemia placentaria(45). Agentes farmacológicos como las estatinas estimulan la expresión de HO-1 e inhiben la liberación de sFlt-1, y potencialmente podrían tener un impacto positivo en el manejo de la preeclampsia que se presenta en el embarazo temprano.
Fetuína-A
La fetuína-A humana, conocida también como glicoproteína Heremans-Schmid alfa-2, es una glicoproteína circulante producida en cantidades elevadas en la vida fetal por varios tejidos, principalmente el hígado(46). Niveles elevados de fetuína-A han sido observados en pacientes con resistencia a la insulina y síndrome metabólico(47,48). Se sabe que esta glicoproteína inhibe la actividad tirosina quinasa del receptor de insulina(49-51). Como muchos factores de crecimiento que promueven la migración del trofoblasto en la vasculatura uterina materna, se unen a receptores que activan la tirosina quinasa(52-56). Nuestro grupo de trabajo lanzó la hipótesis de que la fetuína- A inhibiría la actividad tirosina quinasa de los factores de crecimiento trofoblástico afectando la migración en el útero, resultando en una placentación defectuosa como la observada en preeclampsia(57). Utilizando modelos in vitro de células humanas del trofoblasto extravelloso, demostramos que niveles incrementados de fetuína- A afectaban la invasión trofoblástica, inclusive en presencia de factores estimuladores del crecimiento trofoblástico(57). Además, nuestro grupo demostró que el suero de pacientes embarazadas afectadas por preeclampsia tenía niveles más elevados de fetuína-A comparadas con pacientes que no desarrollaron preeclampsia(57).
Es posible que en un subgrupo de pacientes, las concentraciones elevadas de fetuína-A limiten la migración fisiológica del trofoblasto en la vasculatura uterina, predisponiendo al desarrollo de preeclampsia. Es posible también que estas pacientes con niveles elevados de fetuína-A que tuvieron preeclampsia en el embarazo presenten síndrome metabólico a largo plazo(58).
Daño en los podocitos
La proteinuria relacionada a la preeclampsia se debe al edema de la célula endotelial y a la disrupción del endotelio fenestrado en el podocito renal. Estudios en humanos han demostrado que la expresión de proteínas podocito-específicas está afectada severamente en la preeclampsia. Se ha encontrado expresión disminuida de las proteínas podocito-específicas nefrina, proteína glomerular epitelial 1 (GLEPP-1) y ezrina en secciones de tejido renal en mujeres con preeclampsia comparado con pacientes con presión normal o con hipertensión crónica pregestacional(59). Se ha observado la expresión disminuida de nefrina y sinaptopodina en tejido renal de pacientes que murieron debido a complicaciones de la preeclampsia, comparada con pacientes con presión normal que fallecieron por otras causas no relacionadas a la preeclampsia(60). La detección de podocitos y de productos podocitarios en orina (podocituria) sugiere que la patología relacionada al podocito es más seria de lo anticipada(61-66).
La detección de podocina por métodos de tinción es más sensible y específica en el diagnóstico de preeclampsia al momento del parto que el uso de sinaptopodina, nefrina y pococalixina(61). La podocituria aparece antes del inicio de proteinuria, y el número de podocitos se correlaciona directamente con el grado de proteinuria, lo que sugiere una relación causa-efecto entre la continua pérdida de podocitos y el inicio y severidad de la proteinuria(67).
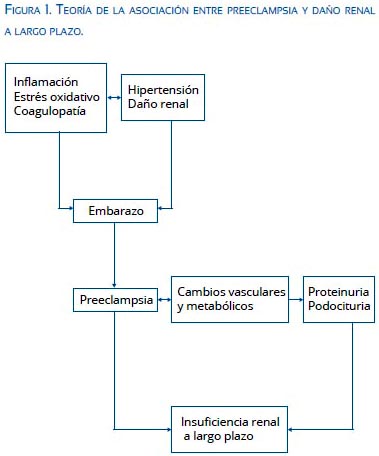
Además de la utilidad de los podocitos como marcadores diagnósticos en preeclampsia, el daño a nivel de los podocitos afectaría la función renal a largo plazo en pacientes con historia de preeclampsia. Estas pacientes tienen un riesgo incrementado de albuminuria, daño renal crónico, y enfermedad terminal (figura 1)(68-71). La podocituria se observa en pacientes con esclerosis glomerular focal segmentaria; esta lesión de esclerosis glomerular es similar a la que se presenta en mujeres con proteinuria persistente después de la preeclampsia(72).
Infecciones durante el embarazo
La infección temprana en el embarazo con el virus adeno-asociado tipo 2 (AAV-2), miembro de la familia de los parvovirus, induce disfunción placentaria(73). Midiendo anticuerpos IgM en el primer trimestre en una muestra aleatoria de pacientes embarazadas, nuestro grupo de trabajo demostró que los niveles de IgM contra AAV-2 se encontraban muy elevados en pacientes que luego desarrollaron preeclampsia, restricción del crecimiento fetal u óbito fetal (eventos obstétricos asociados con disfunción placentaria)(73).
Nuestro grupo también investigó los efectos de la infección por virus del papiloma humano (VPH) de alto riesgo (tipos 16 y 18) en el embarazo. En un modelo in vitro de células trofoblásticas del primer trimestre, la infección por VPH 16 o 18 se asoció con incremento de la apoptosis celular y disminución de la invasión trofoblástica(74). Si bien, la correlación clínica en nuestros experimentos con preeclampsia fue no significativa(74), un estudio retrospectivo reciente encontró que la infección por VPH de riesgo alto duplicaba el riesgo de preeclampsia(75). Actualmente, en la Universidad de Tennessee estamos conduciendo un estudio prospectivo que nos permita evaluar los efectos del VPH 16 y 18 en pacientes en pacientes de riesgo bajo y alto.
En las última dos décadas, numerosos reportes han asociado la infección por Chlamydia pneunomiae con aterosclerosis(76-79). La aterosclerosis se caracteriza también por disfunción endotelial y humoral similar al observado en preeclampsia(80,81). Nuestro grupo de trabajo postuló que la infección aguda por esta bacteria en estadios tempranos del embarazo induciría cambios fisiopatológicos en el trofoblasto y humorales en suero materno similares a los que se presentan en la preeclampsia. Usando modelos in vitro de células humanas del trofoblasto extravelloso, demostramos que Chlamydia pneumoniae era capaz de infectar la placenta, y que esta infección afectaba significativamente el grado de invasión trofoblástica(82). Se encontró el ADN de la bacteria más frecuentemente en placentas de pacientes que desarrollaron preeclampsia(82). En otros experimentos demostramos que pacientes con preeclampsia con manifestaciones severas (preeclampsia severa) presentaban anticuerpos IgG contra Chlamydia pneumoniae, indicativo de infección bacteriana crónica; más aún, los niveles de citoquinas inflamatorias como interleuquina 8, proteína C-reactiva, TNF-alfa, proteína de choque térmico (HSP-60), se encontraron elevados tanto en suero materno de estas pacientes como en el medio celular de trofoblastos infectados con Chlamydia pneumoniae(83). En un reciente trabajo, nuestro grupo encontró que el uso de dosis bajas de aspirina protegía al trofoblasto de los efectos adversos de la infección aguda y crónica por Chlamydia pneumoniae, especialmente en lo que se refiere a la invasión trofoblástica y liberación de mediadores inflamatorios(84). Actualmente, estamos investigando el efecto protector de simvastatina en trofoblastos extravellosos infectados con Chlamydia pneumoniae, usando el mismo modelo in vitro mencionado anteriormente.
CONCLUSIONES
Pese al enorme progreso en la comprensión en la fisiopatología de la preeclampsia en la última década, existen aún muchas preguntas por contestar. El entendimiento de los mecanismos moleculares y celulares involucrados en los distintos estadios de este síndrome ayudará a ofrecer alternativas terapéuticas orientadas a bloquear estos mecanismos y controlar la preeclampsia de manera exitosa.
REFERENCIAS BIBLIOGRÁFICAS
1. Lindheimer MD, Roberts JM, Cunningham GC, Chesley L. En: Lindheimer MD, Roberts JM, Cunningham GC, eds. Chesleys Hypertensive Disorders in Pregnancy. Elsevier, 2009:1-24.
2. Romero R, Lockwood C, Oyarzun E, Hobbins JC. Toxemia: new concepts in an old disease. Semin Perinatol. 1988;12:302-23.
3. Redman, CW, Sargent IL. Latest advances in understanding preeclampsia. Science. 2005;308:1592-94.
4. Roberts JM, Gammill HS. Preeclampsia: recent insights. Hypertension. 2005;46:1243-9.
5. Sibai B, Dekker G, Kupferminc M. Pre-eclampsia. Lancet. 2005;365:785-99.
6. Steegers EA, von Dadelszen P, Duvekot JJ, Pijnenborg R. Preeclampsia. Lancet. 2010 Aug 21;376(9741):631-44. doi: 10.1016/S0140-6736(10)60279-6.
7. American College of Obstetricians and Gynecologists Task Force on Hypertension in Pregnancy 2013. http://www.acog.org/resources_and_publications/task_force_and_work_group_reports/hypertensioninpregnancy (2013).
8. Hutcheon JA, Lisonkova S, Joseph KS. Epidemiology of pre-eclampsia and the other hypertensive disorders of pregnancy. Best Pract Res Clin Obstet Gynaecol. 2011 Aug;25(4):391-403. doi: 10.1016/j. bpobgyn.2011.01.006.
9. Saftlas AF, Olson DR, Franks AL, Atrash HK, Pokras R. Epidemiology of preeclampsia and eclampsia in the United States, 1979-1986. Am J Obstet Gynecol. 1990;163:460-5.
10. Zhang J, Zeisler J, Hatch MC, Berkowitz G. Epidemiology of pregnancy-induced hypertension. Epidemiol Rev. 1997;19:218-32.
11. Eskenazi B, Fenster L,Sidney S. A multivariate analysis of risk factors for preeclampsia. JAMA. 1991;266:237-41.
12. Bodnar LM, Ness RB, Markovic N, Roberts JM. The risk of preeclampsia rises with increasing prepregnancy body mass index. Ann. Epidemiol. 2005;15,475-82.
13. Branch DW, Silver RM, Blackwell JL, Reading JC, Scott JR. Outcome of treated pregnancies in women with antiphospholipid syndrome: an update of the Utah experience. Obstet Gynecol. 1992;80:614-20.
14. Lima F, Khamashta MA, Buchanan NM, Kerslake S, Hunt BJ, Hughes GR. A study of sixty pregnancies in patients with the antiphospholipid syndrome. Clin Exp Rheumatol. 1996 Mar-Apr;14(2):131-6.
15. Dekker G, Robillard PY, Roberts C. The etiology of preeclampsia: the role of the father. J Reprod Immunol. 2011 May;89(2):126-32. doi: 10.1016/j.jri.2010.12.010.
16. Lie RT, Rasmussen S, Irgens LM. Fetal and maternal contributions to risk of pre-eclampsia: population based study. BMJ. 1998;316,1343-7.
17. Conde-Agudelo A, Villar J, Lindheimer M. Maternal infection and risk of preeclampsia: systematic review and metaanalysis. Am J Obstet Gynecol. 2008;198:7-22. doi: 10.1016/j.ajog.2007.07.040.
18. Schieve LA, Handler A, Hershow R, Persky V, Davis F. Urinary tract infection during pregnancy: its association with maternal morbidity and perinatal outcome. Am J Public Health. 1994;84:405-10.
19. Redman CW, Sargent IL. Immunology of pre-eclampsia. Am J Reprod Immunol. 2010 Jun; 63(6):534-43. doi: 10.1111/j.1600-0897.2010.00831.x.
20. Chesley LC, Annitto JE, Cosgrove RA. The familial factor in toxemia of pregnancy. Obstet Gynecol. 1968;32: 303-11.
21. Thornton JG, Macdonald AM. Twin mothers, pregnancy hypertension and pre-eclampsia. Br J Obstet Gynaecol. 1999;106:570-5.
22. Goddard KA, Tromp G, Romero R, Olson JM, Lu Q, Xu Z, Parimi N,et al. Candidate-gene association study of mothers with pre-eclampsia, and their infants, analyzing 775 SNPs in 190 genes. Hum Hered. 2007;63(1):1-16.
23. Ward K, Lindheimer MD. En: Lindheimer MD, Roberts JM, Cunningham GC,m eds. Chesleys Hypertensive Disorders in Pregnancy. Elsevier, 2009: 51-71.
24. Fukui A, Yokota M, Funamizu A, Nakamua R, Fukuhara R, Yamada K,et al. Changes of NK cells in preeclampsia. Am J Reprod Immunol. 2012 Apr;67(4):278-86. doi: 10.1111/j.1600-0897.2012.01120.x.
25. Nelissen EC, van Montfoort AP, Dumoulin JC, Evers JL. Epigenetics and the placenta. Hum Reprod Update. 2011 May-Jun;17(3):397-417. doi: 10.1093/humupd/dmq052.
26. Pijnenborg R, Vercruysse L, Hanssens M. Fetal-maternal conflict, trophoblast invasion, preeclampsia, and the red queen. Hypertens Pregnancy. 2008;27(2):183-96. doi: 10.1080/10641950701826711.
27. LaMarca BD, Gilbert J, Granger JP. Recent progress toward the understanding of the pathophysiology of hypertension during preeclampsia. Hypertension. 2008 Apr;51(4):982-8. doi: 10.1161/HYPERTENSIONAHA.107.108837.
28. Brosens IA, Robertson WB, Dixon HG. The role of the spiral arteries in the pathogenesis of preeclampsia. Obstet Gynecol Annu. 1972;1:177-91.
29. Damsky CH, Fisher SJ. Trophoblast pseudo-vasculogenesis: faking it with endothelial adhesion receptors. Curr Opin Cell Biol. 1998;10(5):660-6.
30. Hunkapiller NM, Gasperowicz M, Kapidzic M, Plaks V, Maltepe E, Kitajewski J, Cross JC, Fisher SJ. A role for Notch signaling in trophoblast endovascular invasion and in the pathogenesis of pre-eclampsia. Development. 2011 Jul;138(14):2987-98. doi: 10.1242/dev.066589.
31. Colucci F, Boulenouar S, Kieckbusch J, Moffett A. How does variability of immune system genes affect placentation? Placenta. 2011 Aug;32(8):539-45. doi: 10.1016/j. placenta.2011.05.001.
32. Gilbert JS, Ryan MJ, LaMarca BB, Sedeek M, Murphy SR, Granger JP. Pathophysiology of hypertension during preeclampsia: linking placental ischemia with endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2008 Feb;294(2):H541-50.
33. Roberts JM, Bodnar LM, Patrick TE, Powers RW. The role of obesity in preeclampsia. Pregnancy Hypertens. 2011 Jan;1(1):6-16.
34. George EM, Granger JP: Endothelin, key mediator of hypertension in preeclampsia. Am J Hypertens. 2011 Sep;24(9):964-9. doi: 10.1038/ajh.2011.99.
35. Thaete LG, Khan S, Synowiec S, Dayton BD, Bauch J, Neerhof MG. Endothelin receptor antagonist has limited access to the fetal compartment during chronic maternal administration late in pregnancy. Life Sci. 2012 Oct 15; 91(13-14):583-6. doi: 10.1016/j.lfs.2012.02.018.
36. Hung TH, Burton GJ. Hypoxia and reoxygenation: a possible mechanism for placental oxidative stress in preeclampsia. Taiwan J Obstet Gynecol. 2006;45(3):189-200.
37. Walsh SW. Maternal-placental interactions of oxidative stress and antioxidants in preeclampsia. Semin Reprod Endocrinol. 1998;16(1):93-104.
38. Roggensack AM, Zhang Y, Davidge ST. Evidence for peroxynitrite formation in the vasculature of women with preeclampsia. Hypertension. 1999;33(1):83-9.
39. Wang A, Rana S, Karumanchi SA. Preeclampsia: the role of angiogenic factors in its pathogenesis. Physiology. 2009;24:147-58.
40. Burton GJ, Yung HW. Endoplasmic reticulum stress in the pathogenesis of early-onset pre-eclampsia. Pregnancy Hypertens. 2011 Jan;1(1-2):72-8.
41. Bainbridge SA, Smith GN. HO in pregnancy. Free Radic Biol Med. 2005 Apr 15;38(8):979-88.
42. Ahmed A, Rahman M, Zhang X, Acevedo CH, Nijjar S, Rushton I, Bussolati B, St John J. Induction of placental heme oxygenase-1 is protective against TNFalpha-induced cytotoxicity and promotes vessel relaxation. Mol Med. 2000 May;6(5):391-409.
43. Wikström AK, Stephansson O, Cnattingius S. Tobacco use during pregnancy and preeclampsia risk: effects of cigarette smoking and snuff. Hypertension. 2010 May;55(5):1254-9. doi: 10.1161/HYPERTENSIONAHA.109.147082.
44. Cudmore M, Ahmad S, Al-Ani B, Fujisawa T, Coxall H, Chudasama K, et al. Negative regulation of soluble Flt-1 and soluble endoglin release by heme oxygenase-1. Circulation. 2007 Apr 3;115(13):1789-97.
45. George EM, Cockrell K, Aranay M, Csongradi E, Stec DE, Granger JP. Induction of heme oxygenase 1 attenuates placental ischemia-induced hypertension. Hypertension. 2011 May;57(5):941-8. doi: 10.1161/HYPERTENSIONAHA.111.169755.
46. Swallow CJ, Partridge EA, Macmillan JC, Tajirian T, Di-Guglielmo GM, Hay K, et al. alpha2HS-glycoprotein, an antagonist of transforming growth factor beta in vivo, inhibits intestinal tumor progression. Cancer Res. 2004 Sep; 64(18):6402-9.
47. Ix JH, Shlipak MG, Brandenburg VM, Ali S, Ketteler M, Whooley MA.Association between human fetuin-A and the metabolic syndrome: data from the Heart and Soul Study. Circulation. 2006 Apr 11;113(14):1760-7.
48. Stefan N, Fritsche A, Weikert C, Boeing H, Joost HG, Häring HU, Schulze MB. Plasma fetuin-A levels and the risk of type 2 diabetes. Diabetes. 2008 Oct;57(10):2762-7. doi: 10.2337/db08-0538.
49. Auberger P, Falquerha L, Contreras JO, Pages G, Le Cam G, Rossi B, Le Cam A. Characterization of a natural inhibitor of the insulin receptor tyrosine kinase: cDNA cloning, purification, and anti-mitogenic activity. Cell. 1989 Aug 25; 58(4):631-40.
50. Rauth G, Pöschke O, Fink E, Eulitz M, Tippmer S, Kellerer M, et al. The nucleotide and partial amino acid sequences of rat fetuin: identity with the natural tyrosine kinase inhibitor of the rat insulin receptor. Eur J Biochem. 1992 Mar 1;204(2):523-9.
51. Goustin AS, Abou-Samra AB. The "thrifty" gene encoding Ahsg/Fetuin-A meets the insulin receptor: insights into the mechanism of insulin resistance. Cell Signal. 2011 Jun;23(6):980-90. doi: 10.1016/j.cellsig.2010.11.003.
52. Fowden AL, Forhead AJ. Endocrine regulation of fe-to-placental growth. Horm Res. 2009;72(5):257-65. doi: 10.1159/000245927.
53. Forbes K, Westwood M. Maternal growth factor regulation of human placental development and fetal growth. J Endocrinol. 2010 Oct;207(1):1-16. doi: 10.1677/JOE-100174.
54. Hashimoto R, Sakai K, Matsumoto H, Iwashita M. Tumor necrosis factor-alpha (TNF-alpha) inhibits insulin-like growth factor-I (IGF-I) activities in human trophoblast cell cultures through IGF-I/insulin hybrid receptors. Endocr J. 2010;57(3):193-200.
55. Giaccone G, Wang Y. Strategies for overcoming resistance to EGFR family tyrosine kinase inhibitors. Cancer Treat Rev. 2011 Oct;37(6):456-64. doi: 10.1016/j. ctrv.2011.01.003.
56. Street ME, Viani I, Ziveri MA, Volta C, Smerieri A, Bernasconi S. Impairment of insulin receptor signal transduction in placentas of intra-uterine growth-restricted newborns and its relationship with fetal growth. Eur J Endocrinol. 2011 Jan;164(1):45-52. doi: 10.1530/EJE-100752.
57. Gomez LM, Anton L, Srinivas SK, Elovitz MA, Parry S. Effects of increased fetuin-A in human trophoblast cells and associated pregnancy outcomes. Am J Obstet Gynecol. 2012 Dec; 207(6):484.e1-8. doi: 10.1016/j.ajog.2012.10.872.
58. Gómez LM. Understanding the effects of fetuin-A in pregnancy. Med J Obstet Gynecol. 2013 Aug 31;1(2):1010.
59. Zhao S, Gu X, Groome LJ, Wang Y. Decreased nephrin and GLEPP-1, but increased VEGF, Flt-1, and nitrotyrosine, expressions in kidney tissue sections from women with preeclampsia. Reprod Sci. 2009 Oct;16(10):970-9. doi: 10.1177/1933719109338630.
60. Garovic VD, Wagner SJ, Petrovic LM, Gray CE, Hall P, Sugimoto H, Kalluri R, Grande JP. Glomerular expression of nephrin and synaptopodin, but not podocin, is decreased in kidney sections from women with preeclampsia. Nephrol Dial Transplant. 2007 Apr; 22(4):1136-43.
61. Garovic VD, Wagner SJ, Turner ST , Rosenthal DW, Watson WJ, Brost BC, et al. Urinary podocyte excretion as a marker for preeclampsia. Am J Obstet Gynecol. 2007 Apr;196(4):e321-e327.
62. Aita K, Etoh M, Hamada H, Yokoyama C, Takahashi A, Suzuki T, Hara M, Nagata M. Acute and transient podocyte loss and proteinuria in preeclampsia. Nephron Clin Pract. 2009;112(2):c65-c70. doi: 10.1159/000213083.
63. Jim B, Jean-Louis P, Qipo A, Garry D, Mian S, Matos T, Provenzano C, Acharya A. Podocyturia as a diagnostic marker for preeclampsia amongst high-risk pregnant patients. J Pregnancy. 2012;2012:984630. doi: 10.1155/2012/984630.
64. Facca TA, Kirsztajn GM, Pereira AR, Moreira SR, Teixeira VP, Nishida SK, Sass N. Renal evaluation in women with preeclampsia. Nephron Extra. 2012 Jan;2(1):125-32. doi: 10.1159/000338271.
65. Chen G, Zhang L, Jin X , Zhou Y, Niu J, Chen J, Gu Y. Effects of angiogenic factors, antagonists, and podocyte injury on development of proteinuria in preeclampsia. Reprod Sci. 2013 May;20(5):579-88. doi: 10.1177/1933719112459227.
66. Son GH, Kwon JY, Lee S , Park J, Kim YJ, Yun B, Park JH. Comparison of serum and urinary nephrin levels between normal pregnancies and severe preeclampsia. Eur J Obstet Gynecol Reprod Biol. 2013 Feb;166(2):139-44. doi: 10.1016/j.ejogrb.2012.10.011.
67. Craici IM, Wagner SJ, Bailey KR , Fitz-Gibbon PD, Wood-Wentz CM, et al. Podocyturia predates proteinuria and clinical features of preeclampsia: longitudinal prospective study. Hypertension. 2013 Jun;61(6):1289-96. doi: 10.1161/HYPERTENSIONAHA.113.01115.
68. Kattah AG, Asad R, Scantlebury DC, Bailey KR, Wiste HJ, Hunt SC, Mosley TH, et al. Hypertension in pregnancy is a risk factor for microalbuminuria later in life. J Clin Hypertens. 2013 Sep;15(9):617-23. doi: 10.1111/jch.12116.
69. Wang IK, Muo CH, Chang YC, Liang CC, Chang CT, Lin SY, et al. Association between hypertensive disorders during pregnancy and end-stage renal disease: a population-based study. CMAJ. 2013 Feb 19;185(3):207-13. doi: 10.1503/cmaj.120230.
70. McDonald SD, Han Z, Walsh MW, Gerstein HC, Devereaux PJ. Kidney disease after preeclampsia: a systematic review and meta-analysis. Am J Kidney Dis. 2010 Jun;55(6):1026-39. doi: 10.1053/j.ajkd.2009.12.036.
71. Vikse BE, Irgens LM, Leivestad T, Skjaerven R, Iversen BM. Preeclampsia and the risk of end-stage renal disease. N Engl J Med. 2008 Aug 21;359(8):800-9. doi: 10.1056/NEJMoa0706790.
72. Heaton JM, Turner DR. Persistent renal damage following pre-eclampsia: a renal biopsy study of 13 patients. J Pathol. 1985;147:121-6.
73. Arechavaleta-Velasco F and Gomez L, Ma Y, Zhao J, Mc-Grath CM, Sammel MD, Nelson DB, Parry S. Adverse reproductive outcomes in urban women with adeno-associated virus-2 infections in early pregnancy. Hum Reprod. 2008 Jan; 23(1):29-36.
74. Gomez LM, Ma Y, Ho C, McGrath CM, Nelson DB, Parry S Placental infection with human papillomavirus is associated with spontaneous preterm delivery. Hum Reprod. 2008 Mar;23(3):709-15. doi: 10.1093/humrep/dem404
75. McDonnold M, Dunn H, Hester A, Pacheco LD, Hankins GD, Saade GR, Costantine MM. High risk human papillomavirus at entry to prenatal care and risk of preeclampsia. Am J Obstet Gynecol. 2014 Feb;210(2):138.e1-5. doi: 10.1016/j.ajog.2013.09.040.
76. Saikku P. Seroepidemiology in Chlamydia pneumoniae-atherosclerosis association. European Heart J. 2002;23:263-4.
77. Wong Y-K, Gallagher PJ, Ward ME. Chlamydia pneumoniae and atherosclerosis. Heart. 1999;81:232-8.
78. Saikku P, Leinonen M, Matilla K, Ekman MR, Nieminen MS, Mäkelä PH, et al. Serological evidence of an association of a novel Chlamydia, TWAR, with chronic coronary heart disease and acute myocardial infarction. Lancet. 1988 Oct;2(5):983-6.
79. Sander D, Winbeck K, Klingelhöfer J, Etgen T, Conrad B. Enhanced progression of early carotid atherosclerosis is related to Chlamydia pneumoniae (Taiwan acute respiratory) seropositivity. Circulation. 2001;103:1390-5.
80. Sander D, Winbeck K, Klingelhofer J, Etgen T, Conrad B. Reduced progression of early carotid atherosclerosis after antibiotic treatment and Chlamydia pneumoniae seropositivity. Circulation. 2002;106:2428-33.
81. Redman CWG, Sacks GP, Sargent IL. Preeclampsia: an excessive maternal inflammatory response to pregnancy. Am J Obstet Gynecol. 1999;180:499-506.
82. Gomez LM, Parry S. Trophoblast infection with Chlamydia pneumoniae and adverse pregnancy outcomes associated with placental dysfunction. Am J Obstet Gynecol. 2009 May;200(5):526.e1-7. doi: 10.1016/j.ajog.2009.03.001.
83. Gomez LM, Srinivas S, Elovitz MA, Parry S. Chronic infection with Chlamydia Pneumoniae and preeclampsia: decreased trophoblast invasion and increased cytokine production in the placenta. Reproductive Sciences. 2010;17(3 Suppl):S835.
84. Gomez LM, Srinivas S, Elovitz, M, Parry S. Placental dysfunction and decreased trophoblast invasion induced by infection with Chlamydia pneumoniae is prevented by low-dose acetyl-salicylic acid. Am J Obstet Gynecol. 2012;206(Suppl 1):S269.
Conflictos de interés: Ninguno.
Correspondencia:
Dr. Luis Martín Gómez Carbajal
Department of Obstetrics and Gynecology, University of Tennessee Health Science Center
853 Jefferson Avenue, Rout E102, Memphis, TN 38163, USA
lgomez2@uthsc.edu

