










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Peruana de Ginecología y Obstetricia
versión On-line ISSN 2304-5132
Rev. peru. ginecol. obstet. vol.65 no.3 Lima jul./dic 2019
http://dx.doi.org/10.31403/rpgo.v66i2193
CASO CLÍNICO
Síndrome de Marfan y embarazo gemelar, presentación de un caso
Marfan syndrome and twin pregnancy. Case report
Carolina García Pumasunta1, Alejandro Mayorga Garcés2, Fausto Viteri Mejía3
1 Posgrado de Ginecología, Pontificia Universidad Católica del Ecuador
2 Especialista en Gastroenterología, Hospital General Docente Ambato, Ecuador
3 Especialista en Ginecología, Hospital GinecoObstétrico Isidro Ayora, Ecuador
ABSTRACT
Marfan syndrome is a hereditary disorder of the connective tissue. Pregnant women who suffer from this syndrome have an increased risk of cardiac complications when the aortic diameter is greater than 40 mm. We present the case of a woman with twin pregnancy and preeclampsia and late diagnosis of Marfan syndrome. We discuss the need for early diagnosis, preconception counseling and multidisciplinary management in this type of pathology.
Key words: Marfan syndrome, Pregnancy, Twin pregnancy.
RESUMEN
El síndrome de Marfan es un trastorno hereditario del tejido conectivo. Aquellas mujeres embarazadas que padecen este síndrome, tienen un riesgo incrementado de complicaciones cardiacas cuando el diámetro aórtico es mayor de 40 mm. Relatamos el caso de una mujer que presentó un embarazo gemelar y preeclampsia, en quien el diagnóstico de síndrome de Marfan se realizó de manera tardía. Se discute la necesidad de un diagnóstico precoz, consejería preconcepcional y un manejo multidisciplinario en este tipo de patología.
Palabras clave. Síndrome de Marfan, Embarazo, Embarazo gemelar.
Introducción
El síndrome de Marfan (SM) es un trastorno hereditario, autosómico dominante, que provoca un desorden sistémico del tejido conectivo. Fue descrito por primera vez por Antoine-Bernard Marfan. Se produce por una mutación del gen codificador de la fibrilina 1, localizado en el cromosoma 15q21. Su incidencia es 2 a 3 casos por cada 10 000 individuos de ambos sexos. Los sistemas más afectados son el esquelético, cardiovascular y ocular(1). Actualmente, para el diagnóstico se utiliza los criterios de Ghent modificados, que incluyen criterios tanto para afectados con antecedentes familiares, como para aquellos que no los tienen(2).
Caso clínico
Paciente de sexo femenino de 22 años de edad, sin antecedentes de importancia, presentó embarazo gemelar de 32,5 semanas. Durante el control médico, se encontró tensión arterial de 152/102 mmHg, insuficiencia venosa periférica en extremidades inferiores y proteinuria +++, por lo que se decidió su traslado a un hospital de mayor complejidad, y tratamiento con sulfato de magnesio, hidralazina y maduración pulmonar con betametasona. El contaje manual de plaquetas fue 87 000. Se decidió terminar el embarazo por cesárea.
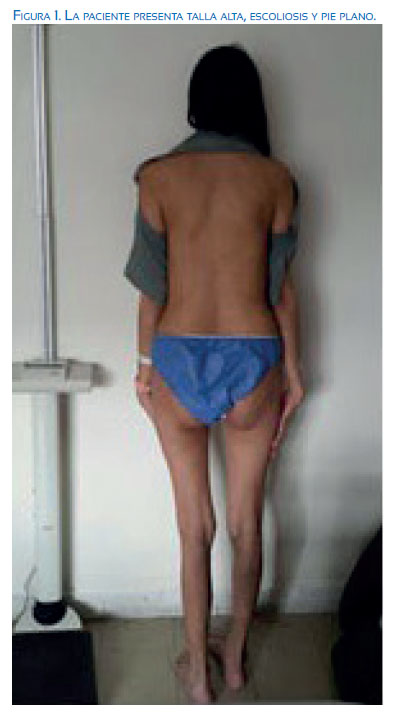
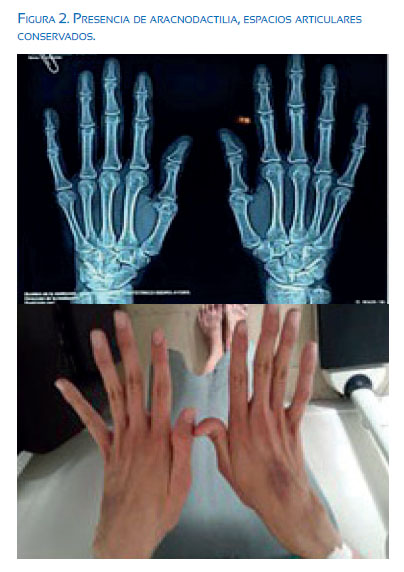
Luego del procedimiento, presentó sangrado aproximado 1 100 mL, sin lograr control con oxitocina y misoprostol, por lo que se realizó histerectomía; permaneció 48 horas en terapia intensiva. La presión arterial se normalizó con uso de amlodipino y atenolol. Al reevaluar el caso, destacaba en el examen físico la talla 181 cm, tórax excavado, signo del pulgar positivo y aracnodactilia (figuras 1 y 2).
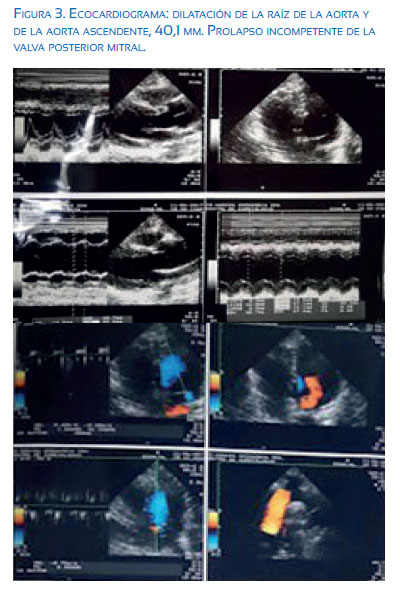
Como antecedentes familiares, dos tíos paternos fueron de talla alta, y fallecieron a los 32 años por cardiopatías no especificadas. El ecocardiograma mostró ausencia de disección o rotura, dilatación de la raíz de la aorta y de la aorta ascendente (figura 3).
La evolución fue satisfactoria, procediéndose al alta y controles en consulta de cirugía cardiaca. Los recién nacidos fueron idénticos, de sexo masculino. Las características fueron las siguientes:
Recién nacido 1: Apgar 7 y 9, peso 1 360 gramos, perímetro cefálico 31 cm, Capurro 33 semanas.
Recién nacido 2: Apgar 7 y 9, peso 1 450 gramos, perímetro cefálico 29 cm, Capurro 33 semanas.
Ninguno presentó signos sugestivos de síndrome de Marfan.
Discusión
Las mujeres afectas con SM generalmente no presentan alteración en la fertilidad; se estima una tasa de hipertensión gestacional de 8%. No se ha demostrado aumento en la incidencia de rotura uterina. El riesgo de partos prematuros es de 6%, restricción en el crecimiento uterino 14% y mortalidad neonatal 1%(3). Actualmente, con los conocimientos sobre la enfermedad y los avances en cirugía cardiovascular, la esperanza de vida es 72 años(4). Las principales causas de mortalidad son la disección y rotura aórtica; la mayoría ocurre en aorta ascendente, siendo más frecuentes en el tercer trimestre o en el posparto temprano(5).
Immer y col determinaron que un diámetro aórtico (DA) mayor de 40 mm y afectación de la válvula bicúspide se asocia con un riesgo incrementado de disección aórtica(6). Esto debido al estado circulatorio hipervolémico e hiperdinámico del embarazo, que conduce a estrés de la pared arterial y cizallamiento de la capa íntima, junto con cambios histológicos producidos por la acción hormonal, que favorecen la dilatación aórtica.
Para el seguimiento, se recomienda un ecocardiograma transtorácico mensual o bimestral para identificar dilatación progresiva, incluso en aquellas con DA menor de 40 mm(7).
En 2010, la Sociedad Americana de Cardiología recomendó la realización de un remplazo profiláctico de la raíz aórtica y la aorta ascendente, cuando el DA es mayor a 40 mm(7).
El uso de betabloqueadores se recomienda en todos los pacientes con SM, pues se asocia con reducción de la dilatación aórtica y el riesgo de disección; los más utilizados son labetalol y metoprolol. No se ha demostrado una relación significativa entre estos y la restricción del crecimiento fetal, pero se recomienda vigilancia durante todo el embarazo.
Los inhibidores de la enzima convertidora de angiotensina (IECA) y los antagonistas de los receptores de angiotensina II (ARA 2) están contraindicados, por su potencial teratógeno. El manejo de los estados hipertensivos no difiere del recomendado en la población general(8).
Se sugiere el parto vaginal en mujeres con SM que tienen un DA menor de 40 mm y no presenten otros factores de alto riesgo. Cuando el DA se encuentra entre 40 y 45 mm, para el parto por vía vaginal se recomienda el uso de anestesia epidural, para disminuir el periodo expulsivo. La cesárea está indicada cuando el DA es mayor de 45 mm y cuando existe regurgitación aórtica o falla cardiaca(9).
El seguimiento posparto debe realizarse por al menos seis semanas, individualizando el riesgo de cada paciente. Se considera contraindicación para el embarazo en quienes presentan un DA mayor de 40 mm, requiriendo una corrección quirúrgica previa, pues se ha demostrado que aquellas con un diámetro aórtico menor a 40 mm tienen un riesgo bajo de complicaciones cardiacas, aproximadamente de 1%(10).
El SM requiere un manejo multidisciplinario, tanto antes como después del embarazo. El equipo médico debe incluir al ginecólogo, neonatólogo, genetista, cardiólogo, cirujano cardiovascular y anestesiólogo. Un diagnóstico adecuado, consejería preconcepcional, vigilancia estricta durante y después del embarazo son claves para la prevención y manejo de las posibles complicaciones del SM, las cuales pueden ser fatales. El uso de betabloqueantes, monitoreo continuo del DA y la cirugía electiva oportuna, permiten alcanzar una sobrevida cercana al 90% (11).
En el caso presentado, la paciente tenía un riesgo incrementado de complicaciones, ya que el SM se diagnosticó de manera tardía. No recibió valoración preconcepcional ni fue evaluada por un equipo multidisciplinario. Además, el embarazo gemelar constituyó un factor de riesgo adicional para otras complicaciones, como parto prematuro, síndrome hipertensivo y restricción del crecimiento fetal. En este caso, el DA se encontraba en el límite superior, y no se presentaron complicaciones cardiacas en la madre ni secuelas en los recién nacidos.
Fuente de financiamiento: autofinanciado
Conflicto de interés: ninguno
Citar como: García Pumasunta C, Mayorga Garcés A, Viteri Mejía F. Síndrome de Marfan y embarazo gemelar, presentación de un caso. Rev Peru Ginecol Obstet. 2019;65(3):345348. DOI: https://doi.org/10.31403/rpgo.v66i2193
Referencias Bibliográficas
1. Bitterman AD, Sponseller PD. Marfan syndrome: A clinical update. J Am Acad Orthop Surg. 2017;25(9):603-609. doi: 10.5435/JAAOS-D-16-00143. [ Links ]
2. Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47(7):476-85. doi: 10.1136/jmg.2009.072785. [ Links ]
3. Donnelly RT, Pinto NM, Kocolas I, Yetman AT. The immediate and long-term impact of pregnancy on aortic growth rate and mortality in women with Marfan syndrome. J Am Coll Cardiol. 2012;60(3):224–9. doi: 10.1016/j.jacc.2012.03.051. [ Links ]
4. Goland S, Elkayam U. Pregnancy and Marfan syndrome. Ann Cardiothorac Surg. 2017;6(6):642-653. doi: 10.21037/acs.2017.10.07. [ Links ]
5. Curry RA, Gelson E, Swan L, Dob D, Babu-Narayan SV, Gatzoulis MA, et al. Marfan syndrome and pregnancy: maternal and neonatal outcomes. BJOG. 2014;121(5):610-7. doi: 10.1111/1471-0528.12515. [ Links ]
6. Immer FF, Bansi AG, Immer-Bansi AS, McDougall J, Zehr KJ, Schaff HV, et al. Aortic dissection in pregnancy: analysis of risk factors and outcome. Ann Thorac Surg. 2003;76(1):309-14. [ Links ]
7. Hiratzka LF, Bakris GL, Beckman JA, Bersin RM, Carr VF, Casey DE Jr, et al. ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with Thoracic Aortic Disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation. 2010;121(13):e266-369. doi: 10.1161/CIR.0b013e3181d4739e. [ Links ]
8. Phipps E, Prasanna D, Brima W, Jim B. Preeclampsia: Updates in pathogenesis, definitions, and Guidelines. Clin J Am Soc Nephrol. 2016;11(6):1102-13. doi: 10.2215/CJN.12081115. [ Links ]
9. European Society of Gynecology (ESG); Association for European Paediatric Cardiology (AEPC); German Society for Gender Medicine (DGesGM), Regitz-Zagrosek V, Blomstrom Lundqvist C, Borghi C, et al. ESC Guidelines on the management of cardiovascular diseases during pregnancy: The Task Force on the Management of Cardiovascular Diseases during Pregnancy of the European Society of Cardiology (ESC). Eur Heart J. 2011;32(24):3147-97. doi: 10.1093/eurheartj/ehr218. [ Links ]
10. Sterner D, Probst C, Mellert F, Schiller W. Surgical treatment and thoracic endovascular aortic repair in type A aortic dissection in a pregnant patient with Marfan syndrome. Ann Vasc Surg. 2014;28(5):1317.e7-10. doi: 10.1016/j.avsg.2013.10.011. [ Links ]
11. Houston L, Tuuli M, Macones G. Marfan syndrome and aortic dissection in pregnancy. Obstet Gynecol. 2011;117(4):956-60. doi: 10.1097/AOG.0b013e3182107310. [ Links ]
Correspondencia:
Dr. Alejandro Mayorga Garcés
Av. Luis Pasteur y Unidad Nacional.
+593 999766562
Recibido: 21 enero 2019
Aceptado: 25 febrero 2019
Publicación online: 22 julio 2019

