










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Peruana de Ginecología y Obstetricia
On-line version ISSN 2304-5132
Rev. peru. ginecol. obstet. vol.65 no.4 Lima Oct./Dic. 2019
http://dx.doi.org/10.31403/rpgo.v65i2216
CASO CLÍNICO
Neurilemoma pélvico benigno degenerado. Reporte de caso
Degenerated benign pelvic neurilemmoma. Case report
Viorkis Pérez-Ortiz1 , Eduardo Reyna-Villasmil2
1 Docente, Facultad de Medicina, Universidad Técnica de Manabí, Portoviejo, Ecuador
2 Médico especialista, Adjunto del Servicio de Ginecología y obstetricia, Hospital Central "Dr. Urquinaona", Maracaibo, Venezuela
ABSTRACT
Neurilemmomas are rare benign tumors derived from the peripheral nerve sheath and composed of perineural Schwann cells proliferating in a characteristic pattern. They are usually benign, slow-growing and often detected incidentally or by local symptoms secondary to compression of neighboring organs. These solitary tumors are located mainly in head and neck, frequently along the path of peripheral nerves, and are particularly rare as retroperitoneal and pelvic tumors. They represent less than 1% of pelvic tumors, and can reach large dimensions. Degenerated or ancient neurilemmomas present typical degenerative characteristics secondary to aging and decreased vascularization. These characteristics can lead to misinterpretation and confusion with a malignant lesion. Tumors are well encapsulated and recurrences are rare following complete surgical excision. The potential risk of surgical excision is neurological injury. We present the case of a degenerated benign pelvic neurilemmoma.
Key words: Neurilemmoma, Schwannomas, Pelvic neoplasms
RESUMEN
Los neurilemomas son tumores raros y benignos derivados de la vaina nerviosa periférica, compuesto por la proliferación de las células de Schwann perineurales con un patrón característico. Generalmente son benignos, de crecimiento lento y, con frecuencia, se les detecta de forma incidental o con síntomas locales secundarios a compresión de órganos vecinos. Estos tumores solitarios se localizan principalmente en cabeza y cuello, a menudo a lo largo del trayecto de los nervios y son particularmente raros entre los tumores retroperitoneales y de la pelvis. Los tumores pélvicos representan menos del 1% de todos los casos, pudiendo alcanzar grandes dimensiones. El neurilemoma degenerado o antiguo presenta características degenerativas típicas secundarias al envejecimiento y disminución de la vascularización. Todas estas características pueden llevar a la interpretación errónea y confundirlo con una lesión maligna. Los tumores están bien encapsulados y las recurrencias después de una escisión quirúrgica completa son poco frecuentes. La escisión quirúrgica conlleva el riesgo potencial de lesión neurológica. Presentamos un caso de neurilemoma pélvico benigno degenerado.
Palabras clave. Paraganglioma vulvovaginal, Feocromocitoma extraadrenal, Genitales femeninos.
Introducción
Los neurilemomas o schwannomas, son neoplasias solitarias benignas, de crecimiento lento, de origen en las células de Schwann y son más comunes en mujeres, entre la segunda y quinta décadas de vida. La transformación maligna es extremadamente rara(1). El neurilemoma degenerado es una variante histológica con amplias porciones compuestas por unas pocas células dentro de una matriz hialinizada. Esta degeneración característica es secundaria a la disminución del flujo sanguíneo en el tiempo a medida que el tumor crece y envejece. La atipia nuclear puede estar presente y confundir con cambios malignos(2,3).
Aunque los neurilemomas retroperitoneales comprenden 0,7% a 2,6% de todos los tumores de este tipo, aquellos de localización pélvica comprenden menos del 1% y la mayoría se origina del plexo hipogástrico o sacro(4). Los neurilemomas pélvicos aparecen como tumores bien encapsulados y vascularizados. En otras ubicaciones miden menos de 5 centímetros de diámetro, pero aquellos con localización pélvica pueden alcanzar tamaños mayores(5). Las dificultades diagnósticas y los retos quirúrgicos de tumores adheridos a la pared y estructuras pélvicas hacen a esta condición difícil de tratar. Se presenta un caso de neurilemoma pélvico benigno degenerado.
Reporte de caso
Se trata de paciente femenina de 69 años quien consultó por presentar aumento de la frecuencia y urgencia miccional acompañado de dolor en fosa iliaca derecha que se extendía hasta la cara anterior del muslo, de aproximadamente tres meses de evolución. Negaba pérdida de peso, alteraciones en hábito o frecuencia evacuatoria, sangrado poscoital y/o flujo o sangrado vaginal; refería inicio de la menopausia natural hace 20 años. También negaba antecedentes personales o familiares de neoplasias malignas.
Durante el examen físico abdominal, se encontró tumoración de consistencia blanda de aproximadamente 15 centímetros, cuyo origen aparente era la pelvis derecha y se extendía hasta el abdomen. El examen vaginal confirmó que la tumoración se originaba aparentemente de la pelvis, la cual también era palpable al tacto rectal. La evaluación neurológica de los miembros inferiores fue normal.
La ecografía abdominal mostró en hemipelvis derecha tumoración quística, redondeada, bien definida y multilocular, de 16 x 14 x 13 centímetros, con septos internos y paredes gruesas y evidencia de flujo sanguíneo capsular y septal por Doppler color. Esto fue confirmado por tomografía computada abdomino-pélvica, mostrando que se extendía hasta el polo inferior del riñón derecho y con atenuación variable, sin evidencia de metástasis ganglionares, peritoneales o a distancia. La vejiga urinaria y el útero estaban desplazados hacia la izquierda, sin visualización del anexo derecho. Las imágenes de resonancia magnética mostraron características del tumor con baja intensidad y márgenes suaves en la imagen ponderada en T1, y de alta intensidad en la imagen ponderada en T2. También se observó hidronefrosis derecha moderada secundaria a la compresión extrínseca del uréter derecho. El hígado, bazo, páncreas y glándulas suprarrenales estaban normales, sin linfadenopatías regionales. Debido a la posibilidad de que la tumoración fuera ovárica maligna, se determinó las concentraciones de marcadores tumorales (antígeno carcinoembriogénico, CA19-9, CA125 y gonadotropina coriónica), los cuales estuvieron dentro de límites normales. En vista de los hallazgos, se decidió programar a la paciente para cirugía.
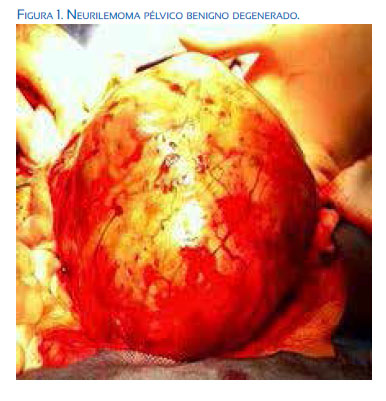
Durante la laparotomía se observó tumoración ovalada y de superficie lisa que medía 20 x 16 centímetros adherida tanto a asas intestinales como a la pared lateral de la pelvis; desplazaba al útero, vejiga urinaria y vasos pélvicos. La tumoración fue cuidadosamente extirpada para minimizar el traumatismo de los tejidos subyacentes. No obstante, se produjo lesión vascular de las venas iliacas comunes e internas, que fue controlada exitosamente con compresión abdominal y sutura de los vasos. Se realizó histerectomía abdominal total, salpingo-ooforectomía bilateral, omentectomía parcial y resección total de la lesión. La pérdida de sangre se calculó en 3 litros, por lo que se utilizó hemoderivados durante la cirugía para mantener las condiciones de la paciente y evitar la coagulopatía. Luego de la cirugía, la paciente fue transferida a la unidad de cuidados intensivos, en donde permaneció 72 horas, y luego fue transferida a hospitalización, siendo dada de alta a los 7 días. En la evaluación postoperatoria, no se encontró deficiencia neurológica en los miembros inferiores.
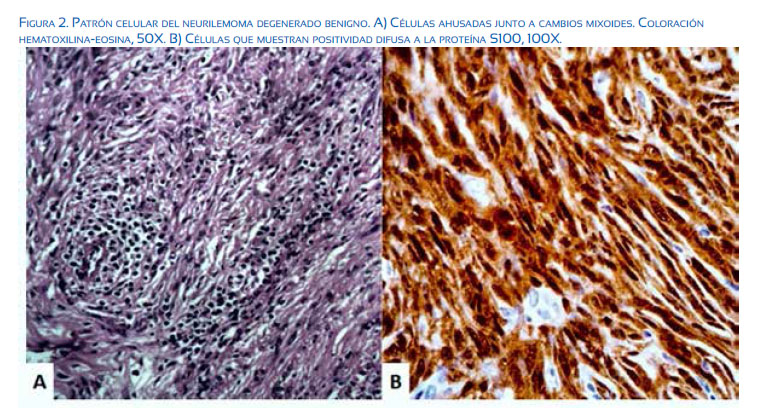
El estudio histopatológico del útero mostró pequeños leiomiomas submucosos en la superficie posterior; ambos ovarios fueron normales. La tumoración pesaba 296 gramos y media 19 x 16 x 14 centímetros, con componentes sólidos y quísticos, color rojo pardusco, con estructuras centrales de aspecto hemorrágico y friable. El examen microscópico mostró algunas pequeñas zonas bien circunscritas compuestas por células ahusadas dispuestas en patrón Antoni tipo A, rodeadas de estructuras eosinofílicas en disposición de organoide (cuerpos de Verocay), junto a otro patrón celular irregular de células fusiformes sin empalmar, en patrón Antoni B. Las células fusiformes mostraron algunas características atípicas, como pleomorfismo celular y atipias nucleares, pero sin alteraciones mitóticas. También se observó estroma vascular con vasos sanguíneos dilatados, áreas focales de degeneración mixoide, junto a otras áreas de necrosis, calcificación y osificación. El análisis inmunohistoquímico para S-100 fue positivo. La coloración inmunohistoquímica de las células ahusadas fue negativa para marcadores epiteliales generales, actina de músculo liso, desmina, calretinina, inhibina, CD10, CK7 y CK20. Estos hallazgos fueron consistentes con el diagnóstico de neurilemoma pélvico benigno degenerado.
La paciente completó 24 meses de seguimiento permaneciendo clínicamente asintomática y libre de enfermedad, desde el punto de vista radiológico.
Discusión
Los neurilemomas pueden aparecer en cualquier nervio mielinizado. Los sitios de origen más comunes son cabeza, cuello y extremidades(2). Generalmente están encapsulados, solitarios y tienen un curso benigno. Pueden variar desde tumores firmes y sólidos hasta quistes fluctuantes. Debido a su crecimiento lento, a menudo se encuentran de forma accidental(3). En algunos casos, su aparición está relacionada con la radioterapia, pero otros se producen en asociación con neurofibromatosis (20% de los casos) o en forma esporádica(6). La mayoría muestra pérdida de alelos en el cromosoma 22(7).
La clasificación de los neurilemomas incluye 5 variantes: común, celular, plexiforme, epitelial y degenerado o antiguo. Este último es una variante rara, que posee áreas hipercromáticas y atipia nuclear con presencia variable de material mixoide y cambios quísticos degenerativos. Representan 0,8% de todos los tumores de tejidos blandos. Las áreas de degeneración son causadas por insuficiencia vascular a medida que el tumor crece. La mayoría aparece en retroperitoneo y mediastino posterior(8).
El crecimiento de los neurilemomas a menudo es lento e indoloro. Aunque las pacientes con neurilemomas pélvicos suelen estar asintomáticos, algunas se quejan de dolor abdominal, pélvico o lumbar, vago e inespecífico, que está relacionado con la compresión de estructuras vecinas(2). Los síntomas urinarios, como incontinencia urinaria y polaquiuria, son secundarios a compresión vesical(4). Además, los resultados de laboratorio generalmente son inespecíficos y no contribuyen al diagnóstico.
El diagnóstico preoperatorio de estos tumores es difícil, por la falta de características típicas en estudios por imágenes; así, pueden ser confundidas con lesiones ováricas malignas. Sin embargo, estos estudios son útiles para establecer tamaño, ubicación y posible invasión de otras estructuras. La biopsia por aspiración con aguja fina puede ser útil, pero si la cantidad y tipo de tejidos son inadecuados, pueden llevar a confusiones debido al pleomorfismo celular en las áreas degeneradas que puede interpretarse como malignidad(5). Los neurilemomas degenerados pueden confundirse con lesiones quísticas debido a la presencia de áreas degeneradas(9). Son descritos generalmente como tumores de baja densidad, poco definidos y heterogéneos en la tomografía computada, mientras que en la resonancia magnética pueden mostrar hipointensidad en imágenes ponderadas en T1 e hiperintensidad en imágenes ponderadas en T2; esto se observa en 57% de los casos(10). Por lo tanto, solo se puede realizar un diagnóstico preciso a partir de la evaluación del material operatorio(1,4).
El diagnóstico definitivo de los neurilemomas se basa en hallazgos histológicos e inmunohistoquímicos. Desde el punto de vista histológico, los tumores tienen células compactas y dos patrones histológicos. El patrón Antoni tipo A, que se caracteriza por disposición centrífuga de células fusiformes y cuerpos de Verocay, mientras que el Antoni tipo B se caracteriza por falta de celularidad tisular y mixoide con células fusiformes muy espaciadas(11). La histopatología del neurilemoma degenerado muestra pérdida relativa de áreas Antoni tipo A, con núcleos irregulares y áreas de hialinización y cambios degenerativos (pleomorfismo, lobulación e hipercromasia). Aparte de las atipias nucleares, otros cambios asociados al proceso degenerativo incluyen formación de quistes, edema estromal, cambio xantomatoso y fibrosis(12). Todos estos cambios han sido atribuidos a la duración del crecimiento del tumor y la consiguiente insuficiencia vascular; de ahí el término ‘degenerado’ o ‘antiguo’. A pesar de dichos cambios, estos tumores se comportan de forma similar a las otras variantes. La diferenciación de la malignidad se puede hacer por la ausencia de mitosis y preservación de grupos cohesivos de células ahusadas(11). Además, casi todas las células muestran tinción inmunohistoquímica intensa para la proteína S-100, lo que confirma el origen neuro-ectodérmico de las células tumorales(13). La citometría de flujo también puede ayudar a confirmar la naturaleza benigna de estos tumores(14).
La diferenciación preoperatoria entre neurofibroma y neurilemoma es fundamental. Los neurofibromas aparecen en el nervio que separa las fibras nerviosas y, por lo tanto, la resección produce lesión nerviosa irreversible, mientras que los neurilemomas surgen de manera excéntrica en el borde nervioso, por lo que la lesión de la raíz nerviosa puede ser evitada(15).
Debido al crecimiento lento, tasa baja de recurrencia y su naturaleza no invasiva que permitiría considerar el tratamiento conservador del neurilemoma como una opción de tratamiento, la resección quirúrgica radical, con preservación de las estructuras circundantes si es posible, es el tratamiento de elección para estos tumores, ya que responden mal a radio-quimioterapia(9). Además, existe la posibilidad de recurrencia local y cambios malignos a pesar del diagnóstico benigno previo. Sin embargo, los problemas quirúrgicos son causados por el origen del tumor. Los tumores viscerales se producen dentro de la fascia pélvica y, por lo tanto, la resección radical es posible con mínimo riesgo de lesión neurológica o vascular. Pero, los tumores de la vaina nerviosa se producen en la fascia pélvica, muy cerca de vasos y raíces nerviosas, por lo que existe un mayor riesgo de lesión del plexo venoso presacro. La cirugía laparoscópica puede ser una opción quirúrgica, ya que ofrece una visión aumentada del campo quirúrgico(4). El pronóstico general es bueno y la recurrencia es rara.
En conclusión, el neurilemoma pélvico degenerado es un tumor benigno, encapsulado y extremadamente raro. Puede confundirse con tumores malignos debido a la atipia celular y otros cambios degenerativos, y alcanza un gran tamaño en la cavidad pélvica, produciendo compresión, lo cual causa la sintomatología. Es importante tener en cuenta este tumor raro en el diagnóstico diferencial de tumoraciones pélvicas y anexiales. El tratamiento de elección es la resección completa del tumor para evitar recurrencias.
Declaración de aspectos éticos
Reconocimiento de autoría. Los autores declaramos que han realizado aportes a la idea, diseño del estudio, recolección de datos, análisis e interpretación de datos, revisión crítica del contenido intelectual y aprobación final del manuscrito que estamos enviando.
Responsabilidades éticas. Protección de personas. Los autores declaramos que los procedimientosseguidosseconformaronalas normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datos. Los autores declaramos que han seguido los protocolos del Hospital Central "Dr. Urquinaona" sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informado. Los autores hemos obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Financiamiento. Los autores certificamos que no han recibido apoyos financieros, equipos, en personal de trabajo o en especie de personas, instituciones públicas y/o privadas para la realización del estudio.
Citar como: Pérez-Ortiz V, Reyna-Villasmil E. Neurilemoma pélvico benigno degenerado. Reporte de caso. Rev Peru Ginecol Obstet. 2019;65(4):555-559. DOI: https://doi.org/10.31403/rpgo.v65i2216
Referencias Bibliográficas
1. Singh M, Kumar L, Chejara R, Prasad OP, Kolhe Y, Saxena A. Diagnostic dilemma of a rare, giant retroperitoneal schwannoma: a case report and review of literature. Case Rep Oncol Med. 2014;2014:628538. doi: 10.1155/2014/628538. [ Links ]
2. Erickson LA. Posterior mediastinal schwannoma (neurilemmoma). Mayo Clin Proc. 2019;94(3):559-560. doi: 10.1016/j.mayocp.2019.01.019. [ Links ]
3. Holliday AC, Mazloom SE, Coman GC, Kolodney MS, Chavan RN, Grider DJ. Benign benign glandular schwannoma with ancient change. Am J Dermatopathol. 2017;39(4):300303. doi: 10.1097/DAD.0000000000000739. [ Links ]
4. Machairiotis N, Zarogoulidis P, Stylianaki A, Karatrasoglou E, Sotiropoulou G, Floreskou A, et al. Pelvic schwannoma in the right parametrium. Int J Gen Med. 2013;6:123-6. doi: 10.2147/IJGM.S41224. [ Links ]
5. Di Furia M, Salvatorelli A, Della Penna A, Vicentini V, Sista F, Chiominto A, et al. Advantage of laparoscopic resection for pelvic Schwannoma: Case report and review of the literature. Int J Surg Case Rep. 2018;45:38-41. doi: 10.1016/j.ijscr.2018.03.006. [ Links ]
6. Rubinstein AB, Reichenthal E, Borohov H. Radiation-induced schwannomas. Neurosurgery. 1989;24(6):929-32. [ Links ]
7. Bello MJ, de Campos JM, Kusak ME, Vaquero J, Sarasa JL, Pestaña A, et al. Clonal chromosome aberrations in neurinomas. Genes Chromosomes Cancer. 1993;6(4):206-11. [ Links ]
8. Ackerman LV, Taylor FH. Neurogenous tumors within the thorax; a clinicopathological evaluation of forty-eight cases. Cancer. 1951;4(4):669-91. [ Links ]
9. Lee NJ, Hruban RH, Fishman EK. Abdominal schwannomas: review of imaging findings and pathology. Abdom Radiol (NY). 2017;42(7):1864-1870. doi: 10.1007/s00261-017-1088-5. [ Links ]
10. Alventosa Mateu C, Castillo López GA, Albert Anteque- ra C. Schwannoma retroperitoneal. Rev Esp Enferm Dig. 2018;110(9):597. doi: 10.17235/reed.2018.5569/2018. [ Links ]
11. Bamgbose BO, Sato A, Yanagi Y, Hisatomi M, Takeshita Y, Sugianto I, et al. A case of schwannoma of the sub- mandibular region. Open Dent J. 2018;12:12-18. doi: 10.2174/1874210601812010012. [ Links ]
12. Gong S, Hafez-Khayyata S, Xin W. Microcystic/reticular Schwannoma: morphological features causing diagnostic dilemma on fine-needle aspiration cytology. Am J Case Rep. 2014;15:538-42. doi: 10.12659/AJCR.892196. [ Links ]
13. Freitas B, Figueiredo R, Carrerette F, Acioly MA. Retrope- ritoneoscopic resection of a lumbosacral plexus schwan- noma: Case report and literature review. J Neurol Surg A Cent Eur Neurosurg. 2018;79(3):262-267. doi: 10.1055/s-0037-1608814. [ Links ]
14. Mastoraki A, Toska F, Tsiverdis I, Kyriazi M, Tsagkas A, Da- nias N, et al. Retroperitoneal schwannomas: dilemmas in diagnostic approach and therapeutic management. J Gas- trointest Cancer. 2013;44(4):371-4. doi: 10.1007/s12029-013-9510-x. [ Links ]
15. Fujibuchi T, Miyawaki J, Kidani T, Miura H. Risk factors for neurological complications after operative treatment for schwannomas. J Clin Neurosci. 2017;46:136-140. doi: 10.1016/j.jocn.2017.09.002. [ Links ]
Correspondencia:
Dr. Eduardo Reyna-Villasmil
Hospital Central "Dr. Urquinaona".
Final Av. El Milagro, Maracaibo, Estado Zulia, Venezuela
584162605233
Recibido: 3 abril 2019
Aceptado: 23 mayo 2019
Publicación online: 14 noviembre 2019

