











 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El síndrome de Cantrell (SC) o pentalogía de Cantrell se describió por primera vez en 1958. Involucra múltiples anomalías congénitas, como son el defecto de la pared abdominal anterior en la línea media, hendidura esternal distal, defecto en el diafragma anterior, defecto del pericardio apical con comunicación pericardio-peritoneal y anomalías intracardiacas1.
La prevalencia de este síndrome varía entre 1:65 000-200 000 nacidos vivos; en países desarrollados, se comunica 5,5 por un millón de recién nacidos vivos2. La mayoría de los casos son esporádicos, con predominancia en el sexo masculino, aunque se ha encontrado asociación con herencia dominante ligada al cromosoma X, aneuploidías como trisomía 18 y 21, infección viral, exposición a sustancias tóxicas como aminopropionitrilo y teratógenos como la quinidina, warfarina, talidomida e incluso la deficiencia de vitamina A3. Se postula que el defecto del tabique transversal es el responsable de esta malformación, que ocurre entre los días 14 y 18 de desarrollo embrionario. El defecto tiene lugar en el proceso de migración o fusión de la línea media anterior del cuerpo1,4,5. El amplio espectro de esta enfermedad puede ser reflejo del momento en que se produce el defecto del desarrollo.
Se comunica uno de los pocos casos de pentalogía de Cantrell diagnosticado prenatalmente en nuestro país, con la revisión de la principal literatura en diagnóstico prenatal.
Caso Clínico
Paciente de 39 años, G3P1011, que inició sus controles prenatales a las 17 semanas de edad gestacional. Se realizó ecografía, observándose gestación única activa de 17 semanas y 6 días con ponderado fetal de 172 gramos. El feto mostraba un defecto de la pared abdominal supraumbilical, que comprometía la porción inferior de esternón y por donde protruía el hígado, intestinos y corazón (se observó el corazón y la porción inferior de ambos pulmones en el defecto); la inserción del cordón umbilical era normal (Figuras 1 a 4). El estudio con Doppler evidenció la alteración de la vasculatura y precisó la extensión del defecto (Figuras 3 y 4). Así mismo, se observó defecto bilateral del labio-palatino (Figura 1A), posición anormal en flexión permanente de manos y polidactilia preaxial (Figura 4B). Se hizo el diagnóstico de síndrome de Cantrell (SD), a descartar probable aneuploidía. No se logró evaluación completa del corazón debido a la posición fetal, por lo cual se planificó examen en 4 semanas. La paciente continuó con sus controles prenatales, pero no se realizó las pruebas de genética solicitadas. Tres semanas después ingresó por rotura de membranas, con 20 semanas 6 días de gestación, produciéndose el parto de un óbito fetal el mismo día. Se corroboró los hallazgos prenatales. Al momento del parto se rompió la membrana que cubría el defecto, impresionando una facies sindrómica con implantación baja de orejas, hipertelorismo y defecto bilateral labio-palatino (Figuras 1, 2 y 4), lo cual aumentó la sospecha de aneuploidía. Los familiares no autorizaron la necropsia.

Figura 1 imagen que muestra la correspondencia entre la imagen 3d (derecha) y la anatomía del producto. se observa el amplio defecto de la pared anterior que contiene hígado, intestinos y corazón. Flecha amarilla: defecto labiopalatino bilateral
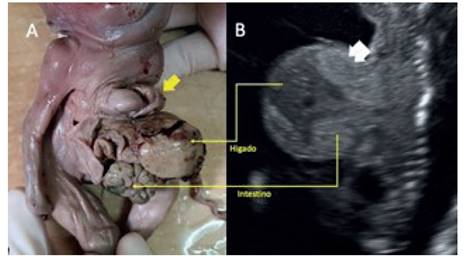
Figura 2 a: imagen del producto con ectopia cordis. flecha amarilla: ventrículo derecho a través de defecto esternal inFerior. b: corte sagital de la ultrasonografía, donde se observa el deFecto abdominal contenido por membrana. Flecha blanca: pulmón fetal dentro del defecto. las flechas amarillas muestran la correspondencia entre las estructuras visibles en el ultrasonido y la patología final.
Discusión
El SC se compone de un defecto en la línea media de la pared abdominal supraumbilical, hendidura esternal distal, defecto del pericardio apical con comunicación pericardio-peritoneal (los dos últimos explican la ectopia cordis), defecto en el diafragma anterior, y anomalías intracardiacas, como defectos del septo ventricular, del septo auriculo-ventricular, tetralogía de Fallot, estenosis pulmonar, entre otros(6,7). En el presente caso, se demostraron todas las características, excepto por la cardiopatía (Figuras 1 a 4), dado que no se pudo realizar la evaluación optima del corazón. No todos los casos expresan las cinco características, por lo que Toyama (1972) sugirió clasificarla por los hallazgos clínicos de la pentalogía, que influyen en el pronóstico y tratamiento de la enfermedad:
Clase 1: diagnóstico exacto; se aprecian los cinco defectos descritos por Cantrell.
Clase 2: diagnóstico probable, con cuatro defectos (que incluyen las anomalías intracardiacas y de la pared abdominal).
Clase 3: diagnóstico incompleto, combinaciones variables de los defectos (siempre incluyendo las anomalías esternales)3).
La ectopia cordis, que se define como la ubicación extra-torácica del corazón, es una de las malformaciones cardíacas asociadas al SC (Figura 3A), pero no es necesaria para el diagnóstico ni está siempre presente.
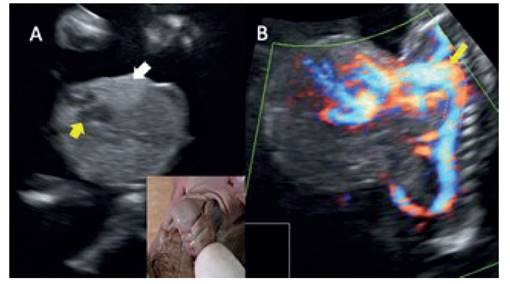
Figura 3 a: corte coronal del defecto; la flecha señala la visión de cuatro cámaras del corazón, zonas hiperecogénicas adyacentes corresponden a pulmones fetales extra-torácicos. recuadro muestra la ectopia cordis del producto. b: corte sagital con uso del power doppler, muestra la silueta de la vascularidad, en la cual se resalta (línea punteada) el cayado aórtico hasta el corazón extratorácico.
El diagnóstico prenatal de la SC debe realizarse idealmente durante el primer trimestre, y confirmar la extensión del compromiso en el segundo, durante los exámenes de rutina. La ultrasonografía volumétrica 3D y el power Doppler nos permiten evaluar con mayor precisión la extensión de la enfermedad y malformaciones asociadas (Figuras 3 y 4). Siempre debe ampliarse con una ecocardiografía, al ser la cardiopatía un determinante del pronóstico. Puede realizarse resonancia magnética fetal en casos no asociados a defectos genéticos, para planificación preoperatoria posnatal2,8-12. Durante el ultrasonido de rutina, la sospecha de ectopia cordis y onfalocele son dos hallazgos ecográficos que deben llevarnos a descartar el SC13. Cuando el defecto es pequeño, la presencia de derrame pericárdico asociado con onfalocele en el primer trimestre requiere confirmación en el segundo trimestre2,7.
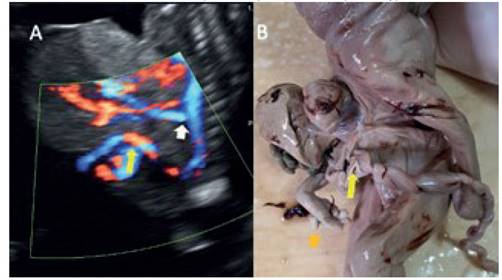
Figura 4 a: corte sagital con power doppler, que muestra los vasos aórticos (flecha blanca: arteria mesentérica) que irrigan los órganos del defecto. flecha amarilla: ingreso de cordón umbilical en la parte baja del defecto. b: imagen del producto, donde se observa el nacimiento del cordón umbilical en la parte inferior del defecto (Flecha amarilla); se observa polidactilia (Flecha naranja).
La variedad de presentación del fenotipo es amplia, tal es así que Sepúlveda14 publica una serie de 7 casos con diagnóstico de ectopia cordis en el primer trimestre, en un periodo de 15 años en tres centros; 4 fueron catalogados como SC, todos caracterizados por el gran defecto en pared abdominal, que contenía hígado y corazón. En cambio, Zidere7 muestra en 3 casos con diagnóstico prenatal temprano que, al llegar al segundo trimestre, la ectopia cordis se había resuelto, pero persistió una efusión pericárdica que puso en evidencia la comunicación entre pericardio y peritoneo; todos los casos se asociaron a cardiopatías severas.
El diagnóstico diferencial incluye ectopia cordis torácica aislada (recordemos que la ectopia cordis puede ser: torácica, toraco-abdominal, abdominal y cervical), síndrome de banda amniótica y anomalía del tallo corporal13. Los estudios genéticos son necesarios por la alta asociación con aneuploidías (trisomía 18, principalmente), que es la principal sospecha en el presente caso. Además, el análisis cromosómico también permite informar y aconsejar mejor a las familias.
El SC con ectopia cordis (EC) se ha asociado con una alta tasa de mortalidad perinatal, al requerir múltiples procedimientos quirúrgicos por etapas15-17. Pośpiech-Gąsio, en ocasión de la publicación de un caso, reúne los casos publicados con diagnóstico prenatal de SC, encontrando 57 casos, en su gran mayoría evaluados en el primer trimestre. De ellos, únicamente 10% sobrevivió, 10% tuvo muerte perinatal y 10% muerte fetal18. Podríamos decir que el pronóstico, en líneas generales, se basa en la presencia de ectopia cordis, la complejidad de la malformación intracardiaca y las malformaciones de otros sistemas asociadas19.
En conclusión, el diagnostico de esta patología debe realizarse en el primer trimestre. La combinación de Doppler color y volumetría 3D mejora aún más la detección del defecto, lo cual nos debe llevar a un asesoramiento oportuno a los padres.

