










Servicios Personalizados
Revista

Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Peruana de Ginecología y Obstetricia
versión On-line ISSN 2304-5132
Rev. peru. ginecol. obstet. vol.68 no.2 Lima abr./jun. 2022 Epub 06-Jul-2022
http://dx.doi.org/10.31403/rpgo.v68i2416
Case report
Prenatal diagnosis of Delleman-Oorthuys syndrome

1. Doctor in Clinical Medicine, Specialist in Obstetrics and Gynecology, Obstetrics and Gynecology Service, Hospital Central "Dr. Urquinaona", Maracaibo, Zulia State, Venezuela.
Delleman-Oorthuys syndrome (oculocerebrocutaneous syndrome) is a rare, congenital, sporadic disorder characterized by microphthalmia/anophthalmia with or without orbital cysts, focal skin defects, and intracranial alterations. Due to an asymmetric distribution of the clinico-radiological features, absence of recurrence within the family and its higher frequency in males, the possibility of somatic mosaicism or sporadic mutations in the fifth or sixth week of fetal development has been proposed. Suggested minimal diagnostic criteria include microphthalmia or orbital cysts, central nervous system cysts or hydrocephalus, and focal skin defects. Given the variability of manifestations and overlap with other syndromes, diagnosis can be difficult, but prenatal detection of this rare congenital anomaly with cerebral malformations is crucial in the management of newborns. Imaging studies for evaluation of features are helpful in the identification and differentiation of cases. A case of prenatal diagnosis of Delleman-Oorthuys syndrome is presented.
Key words: Delleman-Oorthuys syndrome; Eye anomalies; Cutaneous anomalies; Cerebrum anomalies; Prenatal diagnosis.
Introduction
Delleman-Oorthuys syndrome (DOS), or oculocerebrocutaneous syndrome, is a rare, congenital, sporadic oculoneurocutaneous disorder with no known hereditary pattern. It is characterized by periorbital cutaneous appendages, palpebral anomalies, anophthalmia or microphthalmia, orbital cysts, focal alopecia, hypoplastic or aplastic cutis and cerebral alterations such as intracranial cysts and agenesis of the corpus callosum, which is more common in males1. It was first described in 1981 and since then there are reports of about 50 cases, being those diagnosed prenatally very rare2-4. A case of prenatal diagnosis of Delleman-Oorthuys syndrome is presented.
Case report
The patient was 18-year-old, primigravida, who was referred for prenatal high-risk consultation at 26 weeks of gestation due to ultrasound findings of choroid plexus cysts and absence of cerebellar vermis. The patient denied a personal history of seizures, exposure to teratogens, consanguineous ties to her partner, and central nervous system disorders in first-degree relatives.
The ultrasound evaluation performed in the department showed a single female fetus in breech presentation with anterior dorsum, preserved fetal well-being and active fetal movements. The biparietal diameter and amniotic fluid volume were in accordance with gestational age. Three cystic images were observed in the right cerebral hemisphere, the largest measuring 8 x 4 millimeters, deflecting the midline. The largest appeared to extend into the cerebral cortex. The periventricular tissue and cerebral sulci were deformed and displaced by the secondary asymmetric cysts with no evidence of calcifications. No increased echogenicity was observed. The lateral ventricles were normal in size, although deformed, asymmetric, and irregular, with no signs of intraventricular hemorrhage. The third ventricle was normal. Both the corpus callosum and cerebellar vermis were absent and the septum pellucidum was irregular. The retrocerebellar space had a cystic appearance with dilated cisterna magna. Color Doppler ultrasound showed the presence of only the anterior third of the pericallosal artery (Figure 1). The spine ended normally with no abnormalities. The left eyeball was absent, and the right eyeball measured 10 x 7 millimeters.
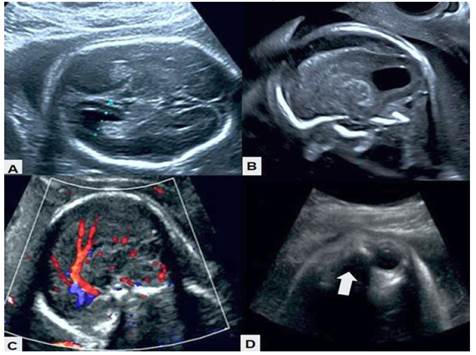
Figure 1 Morphologic ultrasound images. A) Absence of corpus callosum, with deformed and irregular ventricles. B) Asymmetric cyst with deformation and displacement of peripheral tissues. C) Color Doppler in which only the presence of the anterior third of the pericallosal artery is observed. D) The arrow indicates the absence of the left eyeball.
In view of the findings, it was decided to perform fetal magnetic resonance imaging, showing a right paramedian cystic image of approximately 9 x 3 millimeters and agenesis of the corpus callosum. Both cerebral hemispheres were asymmetric, being the left one of greater volume, with areas of lesser thickness and blurred border of the gray-white matter in the parieto-occipital lobes (Figure 2). The basal ganglia could not be differentiated from the white matter because of cerebral asymmetry. The asymmetric lateral ventricles had parallel arrangement with cystic images in the choroid plexuses, one on the right of 9 millimeters and two on the left of 4 and 3 millimeters respectively, with the third ventricle normal. In the posterior fossa, the cerebellum was identified with normal signal and transverse diameter for gestational age, but with a central defect related to the absence of the cerebellar vermis, and a wide cisterna magna with lobulated contours. The brainstem and cerebellum were normal. No intracranial lipomas or cerebral calcifications were observed.
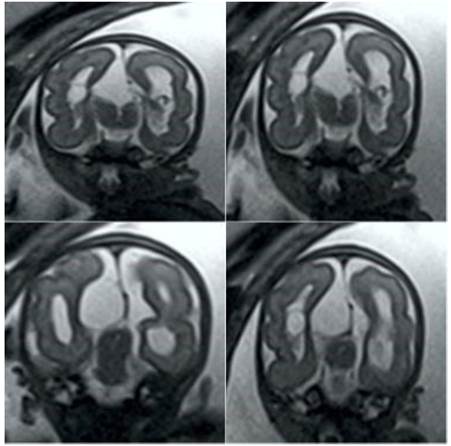
Figure 2 Fetal magnetic resonance imaging sequence showing the asymmetric cerebral hemispheres and lateral ventricles, together with the cystic lesions.
The ocular orbits were of normal size. The left eyeball was noted to be absent, while the right orbit was occupied by a cystic structure whose boundaries were the orbital apex and orbital septum. The fetal profile was normal, although there was protrusion of the skin tissue on the side of the nose that continued with the right eyelid, along with periocular skin lesions near the outer edge of the right orbit. The subcutaneous cellular tissue was well developed in the cervical region. There was no evidence of cleft lip/palate, defects in the spine, thoracic-abdominal organs and extremities. In view of the neurological, cutaneous and ophthalmological findings, the diagnostic possibility of DOS was considered.
The rest of the pregnancy was uneventful, and it was decided to schedule an elective termination of pregnancy at 38 weeks. A live female newborn was obtained in good general condition, with Apgar scores at one minute and 5 minutes of 7 and 9 points, respectively. Initial physical evaluation showed increased volume of the left orbit, without evidence of eyeball, with hypoplastic skin, prominent veins and positive transillumination test, while the left eye was normal. Focal patches of alopecia and dermal hypoplasia of the scalp were also observed, and in the right periocular region there were several mobile cutaneous appendages. Ultrasonography showed fluid-filled cystic areas with no evidence of an eyeball in the right orbit. General examination, including neurologic evaluation, showed no abnormalities and the genitalia, hands and feet were normal. The prenatal diagnosis of Dellerman-Oorthuys was confirmed by postnatal MRI. Both echocardiogram and renal ultrasound showed no abnormalities. The newborn was discharged and referred to plastic surgery, ophthalmology and pediatric neurology consultation for treatment and follow-up.
Discussion
DOS, a rare genetic disorder, is characterized by the presence of cutaneous, ophthalmologic and cerebral alterations. Other more severe forms present cleft or cleft palate and hydrocephalus4. Unilateral onset of manifestations (usually on the left side) is the most common presentation pattern5. In the postnatal period, cutaneous and ocular involvement leads to its identification. Prenatal diagnosis of this condition is usually based on differentiating it from other pathologies3.
The pattern of inheritance is variable, and the specific cause is not known, as most reported cases are sporadic, with no reports of affected siblings5. Some investigators have suggested the possibility of somatic mosaicism or sporadic mutations in the fifth or sixth week of fetal development, affecting the development of midline and midbrain structures, when the midbrain-brain boundaries are established6. The distribution of the characteristic accessory cutaneous appendages along the fusion line of the facial buds suggests alterations in the closure of the embryonic sulci. Similarly, agenesis of the corpus callosum can be considered as a failure of the commissural fibers7,8. Its association to the X chromosome has also been proposed, due to its higher prevalence in males (male:female ratio of 27:7) or because of autosomal dominant inheritance with variable expression9.
The clinical manifestations of DOS are usually related to ocular, cerebral and cutaneous alterations. Possible association with cleft palate, underdeveloped orbit, skull defect, cryptorchidism, complex splenic cyst, micrognathia, rib or vertebral malformation and scoliosis has also been reported3,10. Brain anomalies include more frequent alterations of the frontal lobe, presence of choroidal cysts and malformations of cortical development. There are also reports of tendon reflex changes and positive Babinski's sign on the side opposite the ocular lesion9,11. Neurological abnormalities include seizures, psychomotor developmental disorders (polymicrogyria, colpocephaly, malformations of the ventricular system, hydrocephalus, cystic cavities in the cerebral hemispheres or cerebellum, and agenesis of the corpus callosum). These midline and midbrain malformations are pathognomonic9,12.
Associated ocular anomalies include anophthalmia/microphthalmia (unilateral or bilateral) with or without orbital cysts and iris colobomas1. Orbital cysts have been described as dermoid cysts, orbital encephalocele, cysts with neuroepithelial hamartoma structure, cystic wall surrounded by neuroepithelium and glial tissues or vestigial retinal epithelial tissues11,12. Cutaneous alterations include areas of aplasia or focal hypoplasia, alopecia and formation of the characteristic periocular cutaneous appendages. The cutaneous appendages are pink excrescences in some facial areas, most commonly in the periocular area12.
Neuroimaging studies are key to the diagnosis of DOS. Other evaluations such as electrocardiogram, echocardiography, abdominopelvic ultrasound and spinal radiography are useful to evaluate associated conditions and to rule out/confirm differential diagnoses. Electroencephalogram is useful in those cases presenting with seizures. Prenatal diagnosis with fetal magnetic resonance imaging is very useful1,3. It is not necessary that all features are present to make the diagnosis. Therefore, suggested minimal diagnostic criteria include microphthalmia or orbital cysts, central nervous system cysts or hydrocephalus, and focal skin defects.
The clinical features of DOS may overlap with other established syndromes, which may provide diagnostic dilemmas1. This syndrome shares features with Goldenhar and Goltz-Gorlin syndromes. The former, also known as oculoauriculovertebral syndrome, is characterized by the presence of epibulbar dermoid, preauricular cutaneous appendages, vertebral anomalies and absence of cerebral cysts. The second is an X-linked dominant syndrome, which occurs exclusively in females (in males it is lethal), and may present with microphthalmia, coloboma and focal dermal hypoplasia, in addition to polysyndactyly and deficient dentition12. Overlap with encephalocraniocutaneous lipomatosis or Haberland syndrome has also been described13. In fact, there is some overlap between the two disorders, but this one lacks orbital cysts; and if cutaneous appendages are present, they are not usually located in the periorbital region. In addition, cutaneous hamartomas, epibulbar dermoids and cerebral calcifications are observed. On the other hand, the absence of cerebellar vermis, hypoplastic cerebellar hemispheres and posterior fossa cyst could be confused with Dandy-Walker malformation10,12. However, the coexistence of these anomalies with ocular and cutaneous abnormalities may favor the diagnosis of DOS, which is also more devastating and complex. Other differential diagnoses include Aicardi syndrome, microphthalmia syndrome with linear skin defects and focal dermal hypoplasia12.
Treatment of DOS is multidisciplinary, symptomatic or supportive. Treatment options include removal of the orbital cyst, removal of cutaneous appendages, repair of palpebral lesions, cleft palate, and insertion of ventriculoperitoneal or cystoperitoneal shunt in cases with hydrocephalus1. Treatment of the orbital cyst is aspiration, dissection and removal of the cyst and surrounding periocular structures14. Treatment may involve prolonged anesthesia and extensive dissection, which can be difficult due to brain malformations and associated seizures15.
The prognosis of patients depends on the severity of brain lesions. There are reports of mental retardation in all affected children older than 16 months1. In view of this, detailed neurological evaluations are recommended and, if there is any cerebral or cerebellar manifestation, immediate treatment should be initiated to avoid further neurological damage. Long-term follow-up of these patients is also necessary, as seizures and other neurological manifestations may occur with development12. Ophthalmologic and pediatric evaluations will also be planned in a coordinated manner, for proper management of affected patients.
In conclusion, DOS is a rare congenital disorder characterized by cutaneous, ophthalmologic, and central nervous system lesions. The disorder is possibly caused by sporadic mutations or somatic mosaicism. Prenatal diagnosis is mainly based on suspicion, as some elements may not be found on routine ultrasound evaluation and some features of the syndrome may be confused with other pathologies with similar characteristics.
REFERENCES
1. Bahmani M, Naseri R, Iraniparast A, Mokhtari R, Jafari SH. Oculocerebrocutaneous Syndrome (Delleman Syndrome): A case with a novel presentation of orbital involvement. Case Rep Pediatr. 2021;2021:5524131. doi: 10.1155/2021/5524131 [ Links ]
2. Delleman JW, Oorthuys JW. Orbital cyst in addition to congenital cerebral and focal dermal malformations: a new entity? Clin Genet. 1981;19(3):191-8. doi: 10.1111/j.1399-0004.1981.tb00695.x [ Links ]
3. Brugger PC, Arzt W, Prayer D. Prenatal diagnosis of Delleman syndrome. Prenat Diagn. 2007 Apr;27(4):356-61. doi: 10.1002/pd.1676 [ Links ]
4. Moog U, Dobyns WB. An update on oculocerebrocutaneous (Delleman-Oorthuys) syndrome. Am J Med Genet C Semin Med Genet. 2018;178(4):414-22. doi: 10.1002/ajmg.c.31667 [ Links ]
5. Manudhane A, Arora R, Kapoor S, Rastogi A, Goyal JL. Congenital accessory palpebral aperture--an addition to the spectrum of Delleman syndrome. Ophthalmic Genet. 2013;34(1-2):109-11. doi: 10.3109/13816810.2012.729645 [ Links ]
6. Boycott KM, Dyment DA, Innes AM. Unsolved recognizable patterns of human malformation: Challenges and opportunities. Am J Med Genet C Semin Med Genet. 2018;178(4):382-6. doi: 10.1002/ajmg.c.31665 [ Links ]
7. Ugalahi M, Olusanya B, Fasina O, Seidu M, Adekanmi A. Delleman syndrome: A case report from West Africa - features and the challenges of management. Niger Postgrad Med J. 2018;25(3):191-4. doi: 10.4103/npmj.npmj_75_18 [ Links ]
8. Ortiz-Basso T, Vigo R, Iacouzzi S, Prémoli J. Delleman (oculocerebrocutaneous) syndrome: case report. Indian J Ophthalmol. 2014;62(6):741-3. doi: 10.4103/0301-4738.136277 [ Links ]
9. Divizia MT, Priolo M, Priolo E, Ottonello G, Baban A, Rossi A, et al. How wide is the ocular spectrum of Delleman syndrome? Clin Dysmorphol. 2004;13(1):33-4. doi: 10.1097/00019605-200401000-00009 [ Links ]
10. Saldir M, Polat A, Tunc T, Ozge G, Tehli O, Kacar Y, et al. A newborn with oculocerebrocutaneous syndrome (Delleman Oorthuys syndrome). Genet Couns. 2015;26(4):457-61. [ Links ]
11. Rizvi SW, Siddiqui MA, Khan AA, Siddiqui Z. Delleman Oorthuys syndrome. Middle East Afr J Ophthalmol. 2015;22(1):122-4. doi: 10.4103/0974-9233.148363 [ Links ]
12. Chandravanshi SL, Lakhtakia S. Delleman syndrome or Haberland syndrome? Indian J Dermatol Venereol Leprol. 2014;80(2):155-6. doi: 10.4103/0378-6323.129400 [ Links ]
13. Tian Y, Wang Y, Gao X, Zhang Y, Ju Y. Eye and appearance characteristics of encephalocraniocutaneous lipomatosis. Eye (Lond). 2019;33(2):328-31. doi: 10.1038/s41433-018-0215-z [ Links ]
14. Saatci AO, Arikan G, Saatci P, Saatci Y, Kavukcu S. Oculocerebrocutaneous syndrome. J Pediatr Ophthalmol Strabismus. 2008;45(3):181-3. doi: 10.3928/01913913-20080501-19 [ Links ]
15. Sadhasivam S, Subramaniam R. Delleman syndrome: anesthetic implications. Anesth Analg. 1998;87(3):553-5. doi: 10.1097/00000539-199809000-00009 [ Links ]
Ethical responsibilities: Protection of persons. We the authors declare that the procedures followed conformed to the ethical standards of the responsible human experimentation committee and in accordance with the World Medical Association and the Declaration of Helsinki.
Confidentiality of data: The authors declare that we have followed the protocols of the Ecuadorian Institute of Social Security on the publication of patient data.
Right to privacy and informed consent: The authors have obtained the informed consent of the patients and/or subjects referred to in the article. This document is in the possession of the corresponding author.
Funding: The authors certify that we have not received financial support, equipment, personnel, or in-kind support from individuals, public and/or private institutions for the study.
Received: December 07, 2021; Accepted: March 02, 2022
 Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
