











 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introducción
El síndrome de Delleman-Oorthuys (SDO), o síndrome oculocerebrocutáneo, es un trastorno oculoneurocutáneo raro, congénito y esporádico, sin patrón hereditario conocido. Está caracterizado por apéndices cutáneos periorbitarios, anomalías palpebrales, anoftalmia o microftalmia, quistes orbitarios, alopecia focal, cutis hipoplásico o aplásico y alteraciones cerebrales como quistes intracraneales y agenesia del cuerpo calloso, que es más común en el sexo masculino1. Fue descrito por primera vez en 1981 y desde entonces existen informes de alrededor de 50 casos, siendo los que se diagnostican en el periodo prenatal muy escasos2-4. Se presenta un caso de diagnóstico prenatal del síndrome de Delleman-Oorthuys.
Caso clínico
Se trata de paciente de 18 años, primigesta, quien fue referida a la consulta de alto riesgo prenatal a las 26 semanas de gestación por hallazgos ecográficos de quistes del plexo coroideo y ausencia de vermis cerebeloso. La paciente negaba antecedentes personales de convulsiones, exposición a teratógenos, lazos de consanguinidad con su pareja y alteraciones del sistema nervioso central en familiares de primer grado.
La evaluación ecográfica realizada en el servicio mostró feto único femenino en presentación podálica con dorso anterior, bienestar fetal conservado y movimientos fetales activos. El diámetro biparietal y el volumen de líquido amniótico eran acordes a la edad gestacional. En el hemisferio cerebral derecho se observaron tres imágenes quísticas, la mayor de 8 x 4 milímetros, que desviaban la línea media. El mayor parecía extenderse hasta la corteza cerebral. El tejido periventricular y los surcos cerebrales se encontraban deformados y desplazados por los quistes asimétricos secundarios sin evidencia de calcificaciones. No se observó aumento de la ecogenicidad. Los ventrículos laterales eran de tamaño normal, aunque deformados, asimétricos e irregulares, sin signos de hemorragia intraventricular. El tercer ventrículo estaba normal. Tanto el cuerpo calloso como el vermis cerebeloso estaban ausentes y el septum pellucidum presentaba aspecto irregular. El espacio retrocerebelar tenía aspecto quístico con cisterna magna dilatada. La ecografía Doppler color mostró la presencia solo del tercio anterior de la arteria pericallosa (Figura 1). La espina dorsal terminaba en forma normal sin anomalías. El globo ocular izquierdo estaba ausente y el globo ocular derecho medía 10 x 7 milímetros.
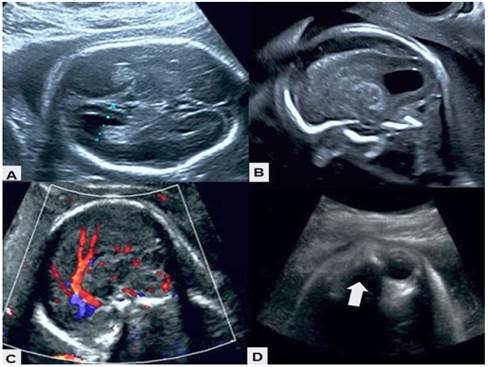
Figura 1 Imágenes de ecografía morfológica. A) Ausencia de cuerpo calloso, con ventrículos deformados e irregulares. B) Quiste asimétrico con deformación y desplazamiento de los tejidos periféricos. C) Doppler color en el cual se observa únicamente la presencia del tercio anterior de la arteria pericallosa. D) La flecha indica la ausencia del globo ocular izquierdo
En vista de los hallazgos se decidió realizar resonancia magnética fetal, observándose imagen quística paramediana derecha de aproximadamente 9 x 3 milímetros y agenesia del cuerpo calloso. Ambos hemisferios cerebrales eran asimétricos, siendo el izquierdo de mayor volumen, con áreas de menor espesor y límite borroso de la materia gris-blanca en los lóbulos parieto-occipitales (Figura 2). Los ganglios basales no pudieron diferenciarse de la sustancia blanca por la asimetría cerebral. Los ventrículos laterales asimétricos tenían disposición paralela con imágenes quísticas en los plexos coroideos, una a la derecha de 9 milímetros y otras dos a la izquierda de 4 y 3 milímetros respectivamente, con el tercer ventrículo normal. En la fosa posterior se identificó el cerebelo con señal y diámetro transverso normal para la edad gestacional, pero con defecto central relacionado a ausencia del vermis cerebeloso, y cisterna magna amplia con contornos lobulados. El tronco encefálico y el cerebelo eran normales. No se observaron lipomas intracraneales ni calcificaciones cerebrales.
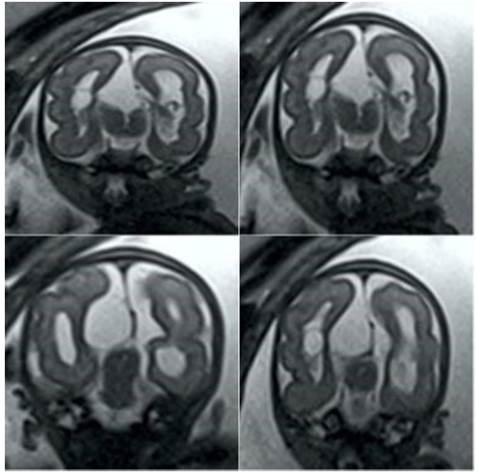
Figura 2 Secuencia de imágenes de resonancia magnética fetal en la que se observan los hemisferios cerebrales y ventrículos laterales asimétricos, junto a las lesiones quísticas.
Las orbitas oculares eran de tamaño normal. Se observó que el glóbulo ocular izquierdo estaba ausente, mientras que la órbita derecha estaba ocupada por una estructura quística cuyos límites eran el vértice orbital y el tabique orbital. El perfil fetal era normal, aunque había protuberancia del tejido cutáneo del lado de la nariz que continuaba con el párpado derecho, junto con lesiones cutáneas perioculares cerca del borde externo de la órbita derecha. El tejido celular subcutáneo estaba bien desarrollado en la región cervical. No se encontraron evidencias de labio / paladar hendido, defectos en la columna vertebral, órganos torácicos-abdominales y en las extremidades. En vista de los hallazgos neurológicos, cutáneos y oftalmológicos, se consideró la posibilidad diagnóstica de SDO.
El resto del embarazo cursó sin problemas y se decidió programar para interrupción electiva del embarazo a las 38 semanas. Se obtuvo recién nacido vivo femenino en buenas condiciones generales, con puntajes de Apgar al minuto y a los 5 minutos de 7 y 9 puntos, respectivamente. La evaluación física inicial mostró aumento de volumen de la órbita izquierda, sin evidencia de globo ocular, con piel hipoplásica, venas prominentes y prueba de transiluminación positiva, mientras que el ojo izquierdo estaba normal. También se observaron parches focales de alopecia e hipoplasia dérmica del cuero cabelludo, y en la región periocular derecha existían varios apéndices cutáneos móviles. La ecografía mostró áreas quísticas llenas de líquido sin evidencia de globo ocular en la órbita derecha. El examen general, incluida la evaluación neurológica, no mostró anormalidades y los genitales, manos y pies eran normales. El diagnóstico prenatal de Dellerman-Oorthuys fue confirmado por la resonancia magnética posnatal. Tanto el ecocardiograma como la ecografía renal no mostraron anomalías. La recién nacida fue dada de alta y referida a la consulta de cirugía plástica, oftalmología y neurología pediátrica para su tratamiento y seguimiento.
Discusión
El SDO, un trastorno genético raro, tiene como característica la presencia de alteraciones cutáneas, oftalmológicas y cerebrales. Otras formas más graves presentan fisura o hendidura del paladar e hidrocefalia4. La aparición unilateral de las manifestaciones (generalmente del lado izquierdo) es el patrón de presentación más común5. En el periodo posnatal, la afección cutánea y ocular conducen a su identificación. El diagnóstico prenatal de esta condición suele basarse en diferenciarlo de otras patologías3.
El patrón de herencia es variable y no se conoce la causa específica, ya que la mayoría de los casos notificados son esporádicos, sin informes de hermanos afectados5. Algunos investigadores han sugerido la posibilidad de mosaicismo somático o mutaciones esporádicas en la quinta o sexta semana de desarrollo fetal, lo que afecta el desarrollo de estructuras de la línea media y mesencéfalo, cuando se establecen los límites entre mesencéfalo y cerebro6. La distribución de los apéndices cutáneos accesorios característicos a lo largo de la línea de fusión de las yemas faciales sugiere alteraciones en el cierre de los surcos embrionarios. Del mismo modo, la agenesia del cuerpo calloso puede considerarse como una falla de las fibras comisurales7,8. También se ha propuesto su asociación al cromosoma X, por su mayor prevalencia en hombres (relación masculino:femenino de 27:7) o como resultado de herencia autosómica dominante con expresión variable9.
Las manifestaciones clínicas del SDO suelen estar relacionadas con alteraciones oculares, cerebrales y cutáneas. También se ha notificado posible asociación con paladar hendido, órbita subdesarrollada, defecto del cráneo, criptorquidia, quiste esplénico complejo, micrognatia, malformación de las costillas o vértebras y escoliosis3,10. Las anomalías cerebrales incluyen alteraciones más frecuentes del lóbulo frontal, presencia de quistes coroidales y malformaciones del desarrollo cortical. También existen informes de modificaciones de los reflejos tendinosos y signo de Babinski positivo en el lado opuesto a la lesión ocular9,11. Las anomalías neurológicas incluyen convulsiones, trastornos del desarrollo psicomotor (polimicrogiria, colpocefalia, malformaciones del sistema ventricular, hidrocefalia, cavidades quísticas en los hemisferios cerebrales o cerebelo y agenesia del cuerpo calloso). Estas malformaciones de línea media y mesencéfalo son patognomónicas9,12.
Entre las anomalías oculares asociadas se incluyen anoftalmia / microftalmia (unilateral o bilateral) con o sin quistes orbitales y colobomas del iris1. Los quistes orbitales han sido descritos como quistes dermoides, encefalocele orbital, quistes con estructura de hamartomas neuroepiteliales, pared quística rodeada por neuroepitelio y tejidos gliales o tejidos epiteliales vestigiales de la retina11,12. Las alteraciones cutáneas incluyen áreas de aplasia o hipoplasia focal, alopecia y formación de los apéndices cutáneos perioculares característicos. Los apéndices cutáneos son excrecencias de color rosado en algunas áreas faciales, más comúnmente en el área periocular12.
Los estudios de neuroimágenes son claves para el diagnóstico del SDO. Otras evaluaciones como electrocardiograma, ecocardiografía, ecografía abdominopélvica y radiografía de columna son útiles para evaluar afecciones asociadas y descartar / confirmar los diagnósticos diferenciales. El electroencefalograma es útil en aquellos casos que presentan convulsiones. El diagnóstico prenatal con imágenes de resonancia magnética fetal es de gran utilidad1,3. Para realizar el diagnóstico no es necesario que todas las características estén presentes. Por lo tanto, entre los criterios de diagnóstico mínimos sugeridos son microftalmia o quistes orbitales, quistes del sistema nervioso central o hidrocefalia y defectos cutáneos focales.
Las características clínicas del SDO pueden superponerse a otros síndromes establecidos, lo que puede ofrecer dilemas diagnósticos1. Este síndrome comparte características con los síndromes de Goldenhar y de Goltz-Gorlin. El primero, también conocido como síndrome oculoauriculovertebral, se caracteriza por presencia de dermoide epibulbar, apéndices cutáneos preauriculares, anomalías vertebrales y ausencia de quistes cerebrales. El segundo es un síndrome dominante ligado a X, que se presenta exclusivamente en mujeres (en hombres es letal), puede cursar con microftalmia, coloboma e hipoplasia dérmica focal, además de polisindactilia y dentición deficiente12. También se ha descrito superposición con la lipomatosis encefalocraniocutánea o síndrome de Haberland13. De hecho, existe cierta coincidencia entre ambos trastornos, pero este carece de quistes orbitales; y si los apéndices cutáneos están presentes, no suelen localizarse en la región periorbital. Además, se observan hamartomas cutáneos, dermoides epibulbares y calcificaciones cerebrales. Por otra parte, la ausencia de vermis cerebeloso, hemisferios cerebelosos hipoplásicos y quiste de la fosa posterior podrían confundirse con la malformación de Dandy-Walker10,12. Sin embargo, la coexistencia de estas anomalías con alteraciones oculares y cutáneas pueden favorecer el diagnóstico del SDO, que, además, es más devastador y complejo. Otros diagnósticos diferenciales incluyen síndrome de Aicardi, síndrome de microftalmia con defectos cutáneos lineales e hipoplasia dérmica focal12.
El tratamiento del SDO es multidisciplinario, sintomático o de apoyo. Las opciones de tratamiento incluyen extirpación del quiste orbital, eliminación de los apéndices cutáneos, reparación de las lesiones palpebrales, paladar hendido e inserción de derivación ventriculoperitoneal o cistoperitoneal en los casos con hidrocefalia1. El tratamiento del quiste orbital es la aspiración, disección y extracción del quiste y estructuras perioculares circundantes14. El tratamiento puede implicar anestesia prolongada y disección extensa, lo cual puede ser difícil por las malformaciones cerebrales y las convulsiones asociadas15.
El pronóstico de los pacientes depende de la severidad de las lesiones cerebrales. Existen informes de retraso mental en todos los niños afectados mayores de 16 meses1. En vista de esto se recomienda realizar evaluaciones neurológicas detalladas y, si existe alguna manifestación cerebral o cerebelosa, iniciar tratamiento inmediato para evitar daños neurológicos adicionales. También es necesario el seguimiento de estos pacientes a largo plazo, ya que pueden aparecer convulsiones y otras manifestaciones neurológicas con el desarrollo12. También se planificará evaluaciones oftalmológicas y pediátricas en forma coordinada, para el manejo adecuado de los pacientes afectados.
En conclusión, el SDO es un desorden congénito raro caracterizado por lesiones cutáneas, oftalmológicas y del sistema nervioso central. El desorden posiblemente es causado por mutaciones esporádicas o mosaicismos somáticos. El diagnóstico prenatal se basa principalmente en la sospecha, ya que algunos elementos pueden no encontrarse en la evaluación ecográfica de rutina y algunas características del síndrome pueden confundirse con otras patologías con características similares.

