











 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
Los tumores hepáticos primarios fetales son poco frecuentes. El hamartoma mesenquimatoso hepático (HMH) es un tumor raro, congénito y benigno, de histogénesis incierta y con pocos casos detectados en el periodo prenatal1,2. Ocupa el segundo lugar en frecuencia, después del hepatoblastoma, cuando se consideran en forma exclusiva los tumores primarios hepáticos3,4. La patogénesis es desconocida y su apariencia ecográfica es variable. La resultante perinatal puede ser potencialmente mortal en el periodo prenatal o neonatal y la importancia de la detección prenatal en el pronóstico aún debe ser establecida2. Se presenta un caso de diagnóstico prenatal de hamartoma mesenquimatoso hepático fetal.
COMUNICACIÓN DEL CASO
Se trata de paciente primigesta de 30 años, con embarazo de 27 semanas por fecha de ultima menstruación, quien fue enviada a la consulta prenatal de alto riesgo por presentar tumor quístico abdominal fetal a las 25 semanas de gestación en la ecografía prenatal de rutina. Las evaluaciones ecográficas realizadas durante el primer y principios del segundo trimestre no revelaron anomalías fetales. La prueba de detección cuádruple a las 16 semanas de gestación mostró elevación de todos los marcadores bioquímicos.
La ecografía en la consulta evidenció feto único de sexo femenino de 27 semanas con biometría acorde a la fecha de ultima menstruación e índice de líquido amniótico que estaba en valores normales para la edad gestacional. En el abdomen fetal se visualizó el hígado aumentado de tamaño con tumor quístico encapsulado en su interior de 28 x 22 milímetros y protuberancias de componentes quísticos-sólidos y claro aspecto anecoico ubicados en el hilio hepático normal por encima de la masa (figura 1). El tumor desplazaba la vena umbilical hacia el lado superior de la vesícula biliar. No se observaron calcificaciones intratumorales. La evaluación Doppler color mostró la naturaleza relativamente avascular del tumor, con flujo Doppler de vena cava inferior y umbilical normal. La placenta no tenía alteraciones ecográficas. Aunque oprimidos por el tumor, los riñones y la vejiga tenían aspecto normal. Las estructuras cardiacas fetales eran anatómicamente normales.
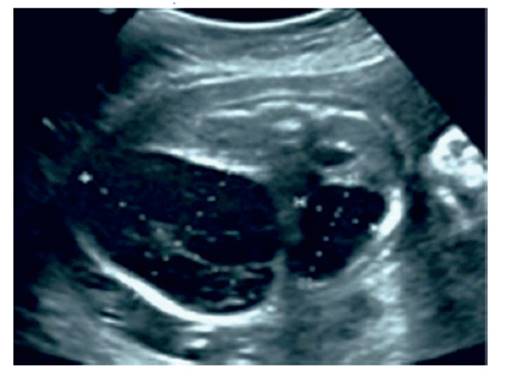
Figura 1 imagen ecográfica transversal del abdomen fetal en la que se observa la tumoración de componentes quísticos-sólidos con claro aspecto anecoico, a las 27 semanas de gestación.
Las imágenes de resonancia magnética fetal a las 28 semanas mostraron que la tumoración era compleja, predominantemente sólida, con áreas quísticas y tabiques internos. No fue detectado algún signo de restricción de difusión. La alta intensidad de señal de la masa en las imágenes ponderadas en T2 (figura 2) con una baja intensidad de señal en las imágenes ponderadas en T1 demostraron marcada hiperintensidad debido a las concentraciones variables de material del quiste. El valor del coeficiente de difusión fue aparentemente de 2,537 mm²/s, por lo que este fue considerado como benigno. El diagnóstico provisional fue el de un HMH fetal. Considerando la edad gestacional, condición fetal y preferencia de los padres fue adoptado el manejo expectante con vigilancia fetal cercana.
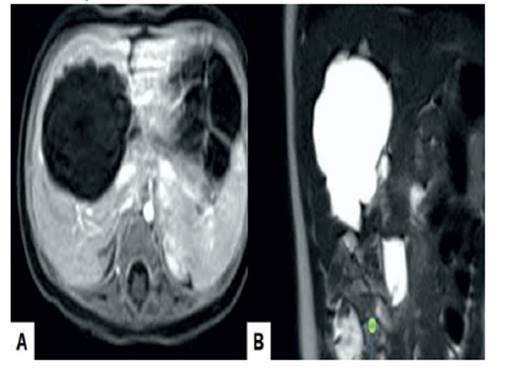
Figura 2 imagen t2 ponderada de resonancia magnética del abdomen fetal que muestra la tumoración hepática fetal.
La evaluación ecográfica a las 33 semanas de gestación reveló que el tumor hepático había aumentado de tamaño (50 x 45 milímetros) con áreas quísticas de mayor tamaño. La circunferencia abdominal fue ubicada en el percentil 99 para la edad gestacional. No se observaron evidencias de polihidramnios, derrame pericárdico, edema subcutáneo generalizado e hidrocele. Se inició el tratamiento prenatal con esteroides para acelerar la maduración pulmonar fetal.
A las 36 semanas se produjo la muerte fetal, por lo que se decidió inducir el parto. Se obtuvo mortinato de sexo femenino de 2,950 gramos por vía vaginal. La evaluación posparto mostró que el abdomen estaba distendido considerablemente, con edema cutáneo generalizado. En la evaluación macroscópica se halló tumor de gran tamaño en el lóbulo hepático derecho, con peso de 600 gramos y una superficie externa lisa con varios quistes de diferentes tamaños, separados por delgadas paredes translúcidas que reemplazaba el tejido hepático (figura 3).
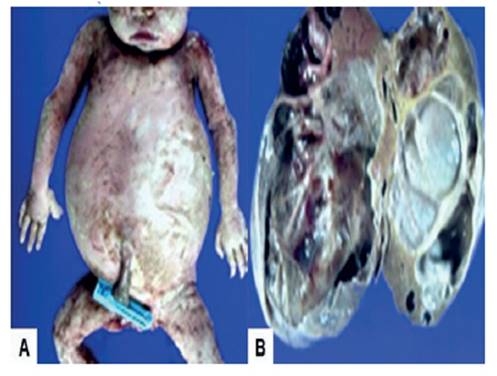
Figura 3 visión macroscópica del tumor. a) feto cuyo abdomen estaba distendido considerablemente con edema cutáneo generalizado. b) imagen macroscópica del hamartoma mesenquimal hepático con patrón quístico predominante y áreas sólidas focales.
La sección del tumor mostró la naturaleza multinodular de color blanco grisáceo, con áreas fibrosas y quísticas ligeramente granulares y septos ocasionales. El quiste mayor medía 15 milímetros y el contenido era amarillento y gelatinoso. El examen histológico señaló que la lesión estaba parcialmente encapsulada y focalmente fusionada con el parénquima hepático circundante. Se encontró proliferación del tejido conectivo mixomatoso con células mesenquimales sueltas en formas de estrella y espacios quísticos intercalados, algunos de los cuales estaban revestidos por el epitelio del conducto biliar. Las porciones sólidas tenían dos elementos principales, uno mixomatoso estromal laxo y otro epitelial, que consistía en nódulos, láminas y cintas de hepatocitos inmaduros y proliferación del conducto biliar a lo largo del lecho del quiste. Este carecía de pleomorfismo, actividad mitótica o focos de hematopoyesis. Los conductos biliares estaban ramificados y eran similares a placas ductales malformadas. El resultado del cariotipo fue 46,XX. El diagnóstico patológico fue de HMH fetal.
DISCUSIÓN
El HMH es un tumor benigno primario caracterizado por crecimiento excesivo de tejidos hepáticos. Son comunes en el período neonatal hasta la infancia tardía, y el 85% de los casos ocurre dentro de los primeros 2 años de vida2. También ha sido descrito como linfangioma, hamartoma de vías biliares, mesenquimoma, tumor mesenquimal pseudoquístico y hamartoma quístico. Generalmente aparece como un tumor asintomático de gran tamaño y rápido crecimiento. El 75% de las lesiones surge en el lóbulo hepático derecho1.
El HMH es una condición de histogénesis poco conocida. La patogenia exacta del hamartoma mesenquimatoso no está clara y tres hipótesis han sido postuladas: anomalía del desarrollo que surge del del mesodermo con desarrollo mesenquimatoso aislado de la arquitectura de la tríada portal normal, un proceso reactivo secundario a la obstrucción del conducto biliar o isquemia regional relacionada con trombosis vascular5,6. Sin embargo, varios estudios han identificado la posible asociación con translocación equilibrada entre los cromosomas 11 19 y rotura en el cromosoma 19q13.4, llevando a la posibilidad de que un subconjunto de estos tumores es verdaderamente neoplásico1. Además, esta última alteración también ha sido identificada en el sarcoma embrionario y el cáncer de ovario. Las calicreínas tisulares (serina proteasas similares a la tripsina) están codificadas por una serie de genes que se localizan en esta región cromosómica. Estas sustancias están involucradas tanto en varias funciones fisiológicas como en el proceso de carcinogénesis7.
El diagnóstico prenatal definitivo de HMH es difícil. La principal característica es de tumor quístico con un componente sólido predominante que aumenta rápidamente de tamaño y resulta en el desplazamiento de los órganos circundantes8. Aunque rara, existen informes de regresión espontánea9. Algunos tumores pueden progresar y causan efectos cardiovasculares y/o alteración del desarrollo pulmonar10. También se le ha asociado con anomalías placentarias como hiperplasia vellosa del tallo mesenquimatoso de la placenta, trombosis o agrandamiento placentario multiquístico(5,11).
Las pruebas de diagnóstico por imágenes más útiles son la ecografía y la tomografía computada6. Desde el punto de vista ecográfico, los hamartomas hepáticos fetales varían en aspecto desde lesiones complejas, multiquísticas y septadas hasta tumor predominantemente sólido. La evaluación Doppler muestra generalmente tumores avasculares, en contraste con el hemangioendotelioma, lo que puede ayudar en el diagnóstico diferencial12. La tomografía computada y la resonancia magnética pueden ser útiles para diferenciar tumores benignos y malignos, ya que puede mostrar las diferentes áreas de atenuación.
Existe evidencia de elevación de las concentraciones séricas de marcadores de aneuploidías, y en especial de alfafetoproteína, en casos de tumores hepáticos fetales. El hepatoblastoma y el carcinoma hepatocelular, junto al HMH, están asociados con mayor producción fetal de alfafetoproteína, lo que explicaría la elevación de las concentraciones maternas por paso transplacentario. También se ha informado elevaciones de la gonadotropina coriónica13. No obstante, las elevaciones de las concentraciones son inespecíficas y están asociadas con varias anomalías congénitas.
Varios tumores entran en los diagnósticos diferenciales del HMH. Los tumores hepáticos benignos pueden dividirse en aquellos de naturaleza neoplásica y no neoplásica. Las lesiones no neoplásicas y neoplásicas de derivación epitelial incluyen quistes simples, enfermedad hepática poliquística, hiperplasias nodulares focales, adenomas y hepatoblastomas3. El hepatoblastoma epitelial-mesenquimatoso mixto es el que es más similar al HMH. Este tipo particular de hepatoblastoma puede estar compuesto por hepatocitos con mesénquima estrellado. El patrón ecogénico heterogéneo es el hallazgo más común, pero algunos casos de hepatoblastoma muestran focos anecoicos debido a necrosis y hemorragia. La resonancia magnética puede mostrar de manera similar imágenes de baja intensidad de señal en T1 y de alta intensidad de señal en T2. El uso de tinciones inmunohistoquímicas puede llevar a confusiones, debido a que las células epiteliales de tipo fetal se tiñen de manera similar a los hepatocitos de tipo adulto (positivo para citoqueratina y hepatocito). Además, los componentes mesenquimales de ambos son positivos para vimentina2.
Los tumores benignos mesenquimales del hígado son más comunes que sus contrapartes epiteliales. Las lesiones vasculares de origen mesenquimatoso son hemangioendotelioma y hemangioma cavernoso. Microscópicamente, estos tumores tienen espacios vasculares variables revestidos por células endoteliales grandes y relativamente inmaduras. El endotelio es positivo para el antígeno relacionado con el factor VIII, CD31, y para las manchas inmunohistoquímicas CD348. Otro diagnóstico diferencial podría ser el neuroblastoma con metástasis hepáticas, el cual puede diagnosticarse en el periodo prenatal por ecografía, resonancia magnética y determinación de las concentraciones de ácido homovanílico en el líquido amniótico14.
El tipo de parto en los casos de HMH debe basarse en indicaciones obstétricas. La vía vaginal es posible en tumores pequeños, pero ya que el tumor puede producir distensión abdominal marcada puede ocurrir rotura traumática y muerte fetal. La cesárea puede considerarse en casos de tumores grandes y riesgo de distocia de tejidos, para reducir el trauma si el feto está vivo12.
El HMH diagnosticado en el periodo prenatal puede estar asociado con una morbimortalidad significativa. Si bien el pronóstico generalmente es considerado bueno en la infancia, la resultante es peor cuando se diagnostica en el período perinatal, con una tasa de mortalidad del 35%11). Puede causar dificultad respiratoria aguda o muerte secundaria a compromiso pulmonar. La descompresión o drenaje prenatal por aspiración guiada por ultrasonido puede aliviar los efectos de masa en los órganos circundantes en tumores grandes con riesgo de compromiso fetal2.
Si bien los HMH generalmente son considerados benignos sin potencial maligno, existe una posible asociación con el sarcoma embrionario indiferenciado del hígado. Ambos son de origen mesenquimatoso y comparten características similares. Aunque es raro, el sarcoma embrionario indiferenciado ocurre dentro del HMH o después de la resección incompleta15).
En conclusión, el HMH es considerado como un tumor congénito, benigno y poco frecuente cuya etiología aún es desconocida. Debido a su presentación inespecífica, puede imitar otras neoplasias benignas y malignas. La apariencia ecográfica es variable y la evaluación Doppler color es útil en el diagnóstico diferencial. El examen histopatológico es fundamental, para poder diferenciarlo de neoplasias malignas. El diagnóstico prenatal está asociado con alta mortalidad, principalmente debido a su efecto de masa.

