











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
A inicios del año 2020, la pandemia de la COVID-19 ha planteado múltiples preocupaciones y ha causado un gran impacto en múltiples áreas de investigación clínica1, específicamente en las disciplinas de biología molecular y virología especializada en epidemiología molecular y genómica2. La aparición de nuevas herramientas genómicas ha sido muy relevante para comprender los distintos aspectos del virus SARS-CoV-23. El análisis genómico completo inicial de la secuencia de dicho virus ha revelado su estado taxonómico como miembro de la familia Betacoronavirus, con una evidente divergencia del SARS-CoV y MERS-CoV4,5.
El análisis genómico incluye tres componentes: secuenciación del ADN, ensamblaje de la secuencia para crear una representación del cromosoma original y anotación y análisis de la representación6. En un análisis filogenético de genomas completos del coronavirus 2 del síndrome respiratorio agudo severo humano (SARS-Cov-2), se encuentran variantes que se distinguen por cambios de aminoácidos. Las muestras se obtienen de casos documentados de enfermedad por COVID-19, lo que indica que las redes filogenéticas también pueden usarse con éxito para ayudar a rastrear fuentes de infección COVID-19, que pueden ponerse en cuarentena para prevenir la propagación recurrente de la enfermedad. en todo el mundo7.
Se pueden utilizar los análisis filogenéticos para dilucidar al paciente reinfectado por SARS-CoV-2 en las que se encuentran dos virus genéticamente distintos en los que se toman las muestras8. Esto debido a las mutaciones entre genomas recientemente reportados en varios momentos y lugares.9. El análisis genómico a gran escala del genoma del SARS-CoV-2 revela la distribución geográfica clonal de las mutaciones delhotspoty la abundante variación genética10. La información sobre la variación de virus tiene un impacto médico y biológico considerable en la prevención, el diagnóstico y la terapia de enfermedades infecciosas9. El objetivo de este manuscrito es describir las secuencias de SARS-CoV-2 aisladas de pacientes peruanos.
METODOLOGÍA
El siguiente estudio es descriptivo observacional, se seleccionaron todos los genomas publicados hasta enero del 2021, se utilizaron datos públicos colgados en el repositorio de GISAID (The Global Initiative on Sharing All Influenza Data) y visualizados mediante la web Nextstrain. Todos los datos están en la web de manera pública, subidas por las mismas instituciones.
La información se filtró por: región, clado GISAID, linaje, y se descargó la información en formato TSV y se creó un árbol filogenético usando la herramienta web iTOL. Las imágenes que se muestran fueron visualizadas en la web Nextstrain usando la forma rectangular de visualización del árbol filogenético y ordenados por fecha.
Se filtró la información por continente, país, región, clado, linaje y sexo desde marzo de 2020 hasta febrero de 2021. Se usó el software estadístico InfoStat para la elaboración de las tablas de frecuencias y la prueba Chi-cuadrado.
RESULTADOS
Se obtuvieron 750 muestras de la secuenciación (no se tiene información del tipo de secuenciamiento) de muestras de SARS-CoV-2 procedentes de Perú, de las cuales 392 pertenecían a pacientes masculinos (52,3 %) y 358 a pacientes femeninas (47,7 %). Además de ello, se confeccionó un árbol filogenético con la aplicacióniTOL v5 Interactive Tree Of Life(Figura 1). Las visualizaciones de los genomas aislados corresponden a las muestras de pacientes del Perú obtenidas desde marzo del 2020 hasta marzo del 2021; además, se filtraron los datos por clado (Figura 2), por región (Figura 3), por linaje viral (Figura 4) y por sexo (Figura 5). Se pueden encontrar las frecuencias en latabla 1.
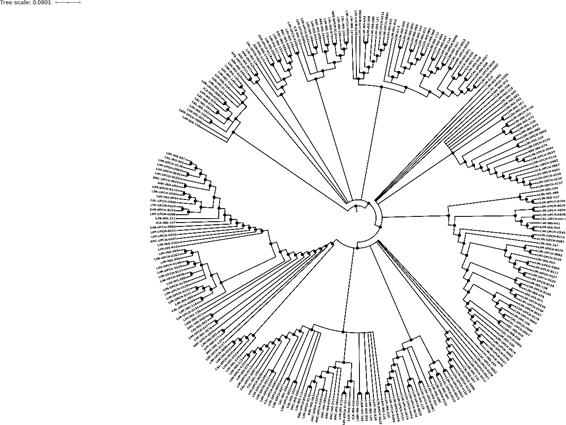
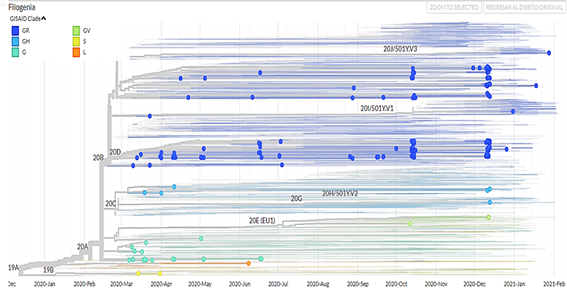
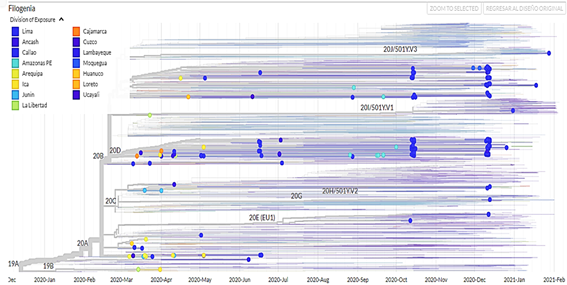
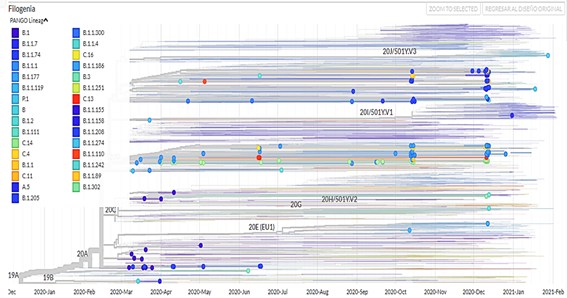
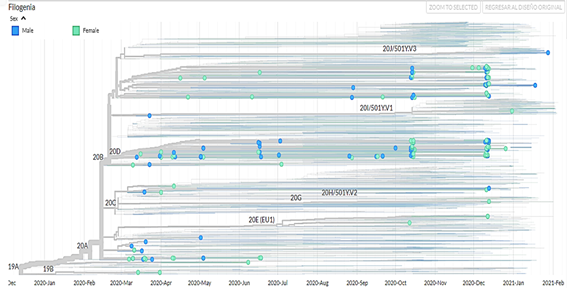
Tabla 1. Frecuencia de linajes y clados por sexo aislados de pacientes de Perú de marzo del 2020 a marzo del 2021.
| Linaje | Femenino | Masculino | Total | % | p |
|---|---|---|---|---|---|
| A | 2 | 0 | 2 | 0.27 | 0.278 |
| A.1 | 2 | 0 | 2 | 0.27 | |
| A.2 | 5 | 1 | 6 | 0.8 | |
| A.5 | 3 | 1 | 4 | 0.53 | |
| B | 1 | 1 | 2 | 0.27 | |
| B.1 | 24 | 28 | 52 | 6.93 | |
| B.1.1 | 87 | 93 | 180 | 24 | |
| B.1.1.1 | 41 | 49 | 90 | 12 | |
| B.1.1.10 | 0 | 2 | 2 | 0.27 | |
| B.1.1.110 | 5 | 7 | 12 | 1.6 | |
| B.1.1.158 | 2 | 3 | 5 | 0.67 | |
| B.1.1.16 | 0 | 1 | 1 | 0.13 | |
| B.1.1.166 | 1 | 0 | 1 | 0.13 | |
| B.1.1.207 | 1 | 0 | 1 | 0.13 | |
| B.1.1.220 | 1 | 3 | 4 | 0.53 | |
| B.1.1.221 | 5 | 4 | 9 | 1.2 | |
| B.1.1.25 | 1 | 0 | 1 | 0.13 | |
| B.1.1.274 | 0 | 1 | 1 | 0.13 | |
| B.1.1.28 | 1 | 0 | 1 | 0.13 | |
| B.1.1.348 | 14 | 13 | 27 | 3.6 | |
| B.1.1.370 | 0 | 1 | 1 | 0.13 | |
| B.1.1.372 | 0 | 1 | 1 | 0.13 | |
| B.1.1.381 | 6 | 6 | 12 | 1.6 | |
| B.1.1.485 | 4 | 6 | 10 | 1.33 | |
| B.1.1.500 | 1 | 1 | 2 | 0.27 | |
| B.1.1.7 | 3 | 0 | 3 | 0.4 | |
| B.1.111 | 1 | 2 | 3 | 0.4 | |
| B.1.13 | 1 | 0 | 1 | 0.13 | |
| B.1.177 | 3 | 1 | 4 | 0.53 | |
| B.1.177.45 | 2 | 0 | 2 | 0.27 | |
| B.1.2 | 4 | 1 | 5 | 0.67 | |
| B.1.205 | 15 | 12 | 27 | 3.6 | |
| B.1.234 | 0 | 2 | 2 | 0.27 | |
| B.1.547 | 0 | 1 | 1 | 0.13 | |
| B.1.596 | 0 | 1 | 1 | 0.13 | |
| B.1.610 | 0 | 4 | 4 | 0.53 | |
| B.1.8 | 0 | 1 | 1 | 0.13 | |
| B.3 | 1 | 0 | 1 | 0.13 | |
| C.11 | 13 | 23 | 36 | 4.8 | |
| C.13 | 7 | 14 | 21 | 2.8 | |
| C.14 | 58 | 52 | 110 | 14.67 | |
| C.22 | 1 | 1 | 2 | 0.27 | |
| C.23 | 0 | 1 | 1 | 0.13 | |
| C.25 | 1 | 6 | 7 | 0.93 | |
| C.32 | 12 | 9 | 21 | 2.8 | |
| C.33 | 5 | 4 | 9 | 1.2 | |
| C.37 | 4 | 3 | 7 | 0.93 | |
| C.4 | 20 | 29 | 49 | 6.53 | |
| N.4 | 0 | 2 | 2 | 0.27 | |
| P.1 | 0 | 1 | 1 | 0.13 | |
| Total | 358 | 392 | 750 | 100 | |
| Clado | Femenino | Masculino | Total | % | p |
| G | 34 | 37 | 71 | 9.47 | 0.0264 |
| GH | 10 | 14 | 24 | 3.2 | |
| GR | 292 | 335 | 627 | 83.6 | |
| GRY | 3 | 0 | 3 | 0.4 | |
| GV | 5 | 1 | 6 | 0.8 | |
| L | 1 | 0 | 1 | 0.13 | |
| O | 1 | 2 | 3 | 0.4 | |
| S | 12 | 2 | 14 | 1.87 | |
| V | 0 | 1 | 1 | 0.13 | |
| Total | 358 | 392 | 750 | 100 |
Tabla 2. Frecuencia de linajes y clados por región aislados de pacientes de Perú de marzo del 2020 a febrero del 2021.
| Linaje | AMA | ANC | APU | ARE | AYA | CAJ | CAL | CUS | HCV | HUA | ICA | JUN | LAL | LAM | LIM | LOR | MOQ | PAS | PIU | SMN | TAC | UCA | Total | p |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | <0.0001 |
| A.1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | |
| A.2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 8 | |
| A.5 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 9 | |
| B | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| B.1 | 0 | 2 | 0 | 8 | 0 | 0 | 5 | 1 | 2 | 0 | 1 | 14 | 0 | 2 | 38 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 74 | |
| B.1.1 | 11 | 4 | 1 | 21 | 1 | 0 | 17 | 1 | 1 | 6 | 9 | 1 | 1 | 3 | 160 | 33 | 2 | 1 | 8 | 4 | 1 | 1 | 287 | |
| B.1.1.1 | 3 | 5 | 0 | 0 | 0 | 0 | 5 | 0 | 0 | 2 | 5 | 1 | 0 | 3 | 85 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 113 | |
| B.1.1.10 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| B.1.1.110 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 12 | |
| B.1.1.158 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | |
| B.1.1.16 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.1.166 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.1.207 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.1.220 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | |
| B.1.1.221 | 0 | 1 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 9 | |
| B.1.1.25 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | |
| B.1.1.274 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.1.28 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| B.1.1.319 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | |
| B.1.1.348 | 0 | 3 | 0 | 2 | 0 | 0 | 3 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 43 | 4 | 2 | 0 | 0 | 0 | 0 | 0 | 60 | |
| B.1.1.370 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.1.372 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| B.1.1.381 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 10 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 13 | |
| B.1.1.398 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.1.434 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.1.485 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 10 | |
| B.1.1.500 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| B.1.1.54 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.1.7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | |
| B.1.111 | 0 | 0 | 0 | 2 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 7 | |
| B.1.13 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.177 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | |
| B.1.177.45 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| B.1.2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | |
| B.1.205 | 0 | 1 | 0 | 1 | 0 | 0 | 4 | 2 | 0 | 0 | 3 | 1 | 0 | 0 | 26 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 38 | |
| B.1.234 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| B.1.243 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.298 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.547 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| B.1.557 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.561 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | |
| B.1.596 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.610 | 0 | 0 | 0 | 2 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 12 | |
| B.1.8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| C.11 | 0 | 6 | 0 | 1 | 0 | 1 | 4 | 0 | 4 | 11 | 3 | 5 | 1 | 3 | 53 | 9 | 0 | 0 | 1 | 0 | 0 | 0 | 102 | |
| C.13 | 0 | 1 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 1 | 2 | 0 | 0 | 1 | 19 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 28 | |
| C.14 | 13 | 22 | 0 | 0 | 3 | 0 | 12 | 1 | 4 | 2 | 2 | 2 | 1 | 25 | 70 | 3 | 0 | 0 | 3 | 1 | 0 | 0 | 164 | |
| C.22 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | |
| C.23 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| C.25 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 8 | |
| C.32 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 31 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 33 | |
| C.33 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 10 | |
| C.37 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 48 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 51 | |
| C.4 | 1 | 0 | 0 | 0 | 0 | 0 | 7 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 57 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 67 | |
| C.8 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | |
| N.4 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| P.1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 3 | 16 | 0 | 0 | 0 | 0 | 0 | 0 | 24 | |
| Total | 31 | 49 | 3 | 42 | 5 | 3 | 77 | 7 | 11 | 38 | 28 | 27 | 5 | 40 | 750 | 74 | 4 | 1 | 18 | 5 | 1 | 1 | 1220 | |
| Clado | AMA | ANC | APU | ARE | AYA | CAJ | CAL | CUS | HCV | HUA | ICA | JUN | LAL | LAM | LIM | LOR | MOQ | PAS | PIU | SMN | TAC | UCA | Total | p |
| G | 0 | 1 | 0 | 11 | 0 | 0 | 9 | 3 | 2 | 2 | 4 | 6 | 0 | 1 | 67 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 108 | <0.0001 |
| GH | 0 | 2 | 0 | 2 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 9 | 0 | 1 | 22 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 39 | |
| GR | 31 | 45 | 3 | 29 | 5 | 2 | 63 | 4 | 9 | 36 | 22 | 12 | 3 | 36 | 632 | 72 | 4 | 1 | 18 | 5 | 1 | 1 | 1034 | |
| GRY | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | |
| GV | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | |
| L | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| O | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | |
| S | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 2 | 0 | 2 | 0 | 16 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 22 | |
| V | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
AMA: Amazonas; ANC: Ancash; APU: Apurímac; ARE: Arequipa, AYA: Ayacucho; CAJ: Cajamarca; CAL: Callao; CUS: Cusco; HCV: Huancavelica; HUA: Huánuco; ICA: Ica; JUN: Junín; LAL: La Libertad; LAM: Lambayeque; LIM: Lima; LOR: Loreto; MOQ: Moquegua; PAS: Pasco; PIU: Piura; SMN: San Martín; TAC: Tacna; UCA: Ucayali.
DISCUSIÓN
El análisis de los distintos genomas ha sido muy útil para rastrear al virus SARS-CoV-2 en distintas regiones, no solo de Perú, sino también de Latinoamérica y el mundo. En cuanto a los clados, Perú ha identificado al clado GR como en todos los países de América del Sur a excepción de Paraguay que no ha identificado genomas de SARS-CoV-2 hasta la actualidad; por otro lado, el clado G es común entre todos los países menos Venezuela y el clado GH en todos menos Bolivia y Uruguay, siendo éstos los más comunes, seguidos por los clados S, L y GV. Otros clados identificados en este continente, pero no en Perú fueron los clados O y V11.
En Uruguay, el análisis genético muestra que los clados S y G (G, GH, GR) dominan, representando más del 90 % de las cepas del virus en nuestro estudio; el resultado fatal de la infección por SARS-CoV-2 está significativamente relacionado con la hipertensión, Insuficiencia renal e ingreso en UCI (FDR <0,01), pero no hay mutaciones en proteínas estructurales o no estructurales como la proteína pico D614G12. Al igual que en Perú, países de Norteamérica como Canadá ha rastreado al clado GR con mayor frecuencia, seguidos de los clados G y GH, y en menor frecuencia los clados S y L, estos últimos fueron identificados en la misma frecuencia en Estados Unidos, no obstante, en este país se ha identificado con más frecuencia al clado GH seguidos de los clados GR y G; en México, no ocurre lo mismo, ya que los clados más frecuentes son GR y G seguido del GH, siendo igual en el caso de los clados S y L siendo muy menores, además de que en esta parte del continente americano se encontraron los clados O y V11. En Europa y Asia al igual que Perú se ha registrado más los clados GR, G y GH y más abajo están los clados S, L y GV, teniendo en cuenta de que en estos continentes se ha realizado mayor número de análisis de genomas al virus SARS-CoV-211.
En cuanto a los linajes virales se encontró diferencias con los genomas aislados en Argentina ya que el linaje encontrado con mayor frecuencia y persistencia fue el B.1 seguido del B.1.1.33 y el B.1.499, lo mismo pasa en Bolivia que a pesar que solo se ha identificado 14 genomas, el linaje B.1 persistió a través del tiempo; en Brasil y Uruguay, los linajes B.1.1.33 y B.1.1.28 estuvieron presentes aproximadamente todo el tiempo que abarco la pandemia en ese país; mientras, en Chile, Colombia y Ecuador los linajes más frecuentes y persistentes son B.1 y B.1.1.74. En Norteamérica, Europa y Asia el linaje mayoritario es B.1 pero a mediados y finales del 2020, hubo un incremento de los linajes B.1.2, B.1.1.7 y B.1.17713, datos que difieren con el presente estudio.
Además de los análisis de genomas encontrados en plataformas web, se debe considerar estudios publicados en revistas, como es el caso de la investigación realizada en Ecuador donde el linaje más dominante en todas las secuencias de ese país fue B.1.1.74, seguidas de B.1 y B.1.1.114, lo cual no coincide con nuestros resultados; por otro lado, el linaje P.1 fue identificado en ciudades de la amazonia brasileña como Manaos15, lo cual se relaciona con la trasmisión de éste en la región Loreto donde fue el linaje más frecuente después de B.1.1. En cuanto a un estudio realizado en Venezuela, los linajes B.1.1 y B.1.111 fueron los más persistentes en distintas provincias16, lo cual no guarda relación con la presente investigación, al igual que los linajes identificados con mayor incidencia en Sudáfrica como lo son C.1, B.1.1.56 y B.1.1.5417.
Para el caso de los linajes por regiones, Lima ha presentado el mayor número de linajes y clados identificados, por lo tanto, se sugiere que presenta el mayor número de contagios en esa ciudad seguida de Callao, Loreto, Áncash y Arequipa, y también se puede decir que la transmisibilidad del virus en esas ciudades es muy elevada.
Cabe resaltar que el linaje B.1.1.7, ha tenido mucha importancia en cuanto a la hospitalización de pacientes de manera grave o en la muerte de los mismos como indica un estudio del Reino Unido, en donde más del 60% de pacientes muestreados, presentaron esas características18, al igual que en Estados Unidos donde la rápida transmisión de este linaje causó un incremento en las defunciones19, lo cual guardaría relación con el presente estudio, ya que la disminuida manifestación de ese linaje en Perú no se expresa en un aumento desmesurado de hospitalizaciones y muertes como ocurrió en el país europeo. Por último, en un estudio hecho en India se ha evidenciado que los clados GR, GH Y G son los más predominantes con más del 70 %20, coincidiendo con nuestros resultados.
Este estudio retrospectivo tiene limitaciones como el pequeño tamaño de la muestra, no se puede tener control sobre la calidad de datos que se sube y el sesgo en la recopilación de datos.
Por tales motivos, es importante realizar una vigilancia epidemiológica genómica ya que esta nos podría avizorar sobre los nuevos cambios en las variantes de SARS-CoV-2 y el uso de esta información para tomar medidas de contención, como nuevas formas de diagnóstico, monitoreo, clínica, etc. Es importante rastrear la manera en cómo se propaga y si las mutaciones tienen importancia clínica en los pacientes infectados. Esto se logra en un trabajo conjunto del estado con los investigadores.
CONCLUSIÓN
El clado GR es común para todos los países sudamericanos, continentes europeos y asiáticos, seguido de los clados G y GH con mayor frecuencia; por otro lado, el linaje viral más persistente en Perú es el B.1.1, siendo este dato no común con otros países. Además, se identificó la mayoría de los linajes en las regiones Lima, Callao, Loreto, Ancash y Arequipa.

