










Serviços Personalizados
Journal

Artigo
 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista de la Facultad de Medicina Humana
versão impressa ISSN 1814-5469versão On-line ISSN 2308-0531
Rev. Fac. Med. Hum. vol.21 no.3 Lima jul./set. 2021
http://dx.doi.org/10.25176/rfmh.v21i3.3712
Original article
Analysis of SARS-CoV-2 genomes samples from Peru
1Faculty of Biological Sciences, Universidad Nacional Pedro Ruiz Gallo. Lambayeque, Peru.
2Universidad Nacional de Jaén. Cajamarca, Peru.
Introduction:
Genomic analysis of samples from documented COVID-19 cases can be used successfully to help track sources of Sars-Cov-2 infection, which can be quarantined to prevent the recurrent spread of the disease around the world.
Methods:
All genomes published up to March 2021, uploaded in the GISAID and Nextstrain repository, were selected. All data is on the web in a public way; In addition, the information was filtered by continent, country, region, clade, lineage, and sex from March 2020 to February 2021.
Results:
It was evidenced that the region with the most isolated genomes was Lima, the most frequent clade is GR, the viral lineage B.1.1 is the most frequent and persistent in time and most of the genomes were isolated from people of the female sex.
Conclusions:
The clade GR is common to all South American countries and the European and Asian continents, followed by clades G and GH with greater frequency; on the other hand, the most persistent viral lineage in Peru is B.1.1, this being not common with other countries.
Keywords: Coronavirus Infections; Genome Viral; Whole Genome Sequencing; Mutation; Peru. (Source: MeSH - NLM)
INTRODUCTION
At the beginning of 2020, the COVID-19 pandemic raised multiple concerns. It caused a great impact in multiple areas of clinical research1, specifically in the disciplines of molecular biology and virology specialized in molecular and genomic epidemiology2. The appearance of new genomic tools has been very relevant to understand the different aspects of the SARS-CoV-2 virus3. The initial complete genomic analysis of the sequence of this virus has revealed its taxonomic status as a member of the Betacoronavirus family, with an evident divergence from SARS-CoV and MERS-CoV4,5.
The genomic analysis includes three components: DNA sequencing, assembling the sequence to create a representation of the original chromosome, and annotating and analyzing the representation6. In a phylogenetic analysis of whole genomes of the human severe acute respiratory syndrome coronavirus 2 (SARS-Cov-2), variants are found that are distinguished by amino acid changes. The samples are obtained from documented cases of COVID-19 disease, indicating that phylogenetic networks can also be used successfully to help track sources of COVID-19 infection, which can be quarantined to prevent the recurrent spread of the disease worldwide7.
Phylogenetic analyzes can be used to elucidate the SARS-CoV-2 reinfected patient in which two genetically different viruses are found from which samples are taken8. This is due to recently reported cross-genome mutations at various times and places.9. Large-scale genomic analysis of the SARS-CoV-2 genome reveals the clonal geographic distribution of mutationshotspotand abundant genetic variation10. Information on virus variation has considerable medical and biological impact on the prevention, diagnosis, and therapy of infectious diseases9. The objective of this manuscript is to describe the SARS-CoV-2 sequences isolated from Peruvian patients.
METHODOLOGY
The following study is descriptive and observational, all genomes published up to January 2021 were selected, public data posted in the GISAID repository (The Global Initiative on Sharing All Influenza Data) and viewed through the Nextstrain website were used. All data is on the web in a public way, uploaded by the same institutions.
The information was filtered by: region, GISAID clade, lineage, and the information was downloaded in TSV format and a phylogenetic tree was created using the iTOL web tool. The images shown were viewed on the Nextstrain website using the rectangular display form of the phylogenetic tree and ordered by date.
The information was filtered by continent, country, region, clade, lineage and gender from March 2020 to February 2021. The statistical software InfoStat was used to prepare the frequency tables and the Chi-square test.
RESULTS
750 sequencing samples were obtained (there is no information on the type of sequencing) of SARS-CoV-2 samples from Peru, of which 392 belonged to male patients (52.3%) and 358 to female patients (47.7%). In addition, a phylogenetic tree was made with theiTOL v5 Interactive Tree Of Life(Figure 1). The visualizations of the isolated genomes correspond to the samples of patients from Peru obtained from March 2020 to March 2021; in addition, the data were filtered by clade (Figure 2), by region (Figure 3), by viral lineage (Figure 4) and by gender (Figure 5). The frequencies can be found intable 1.
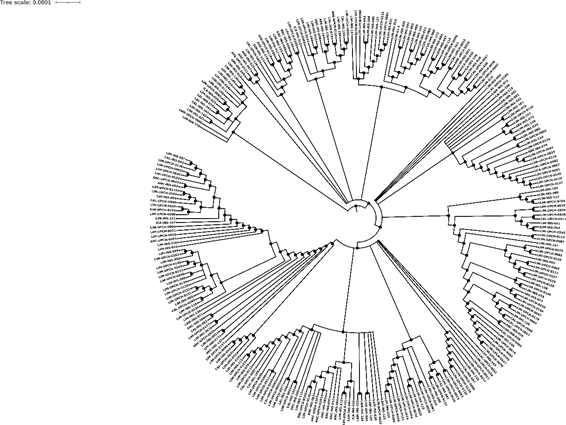
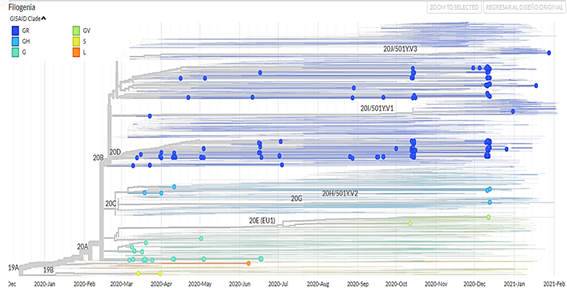
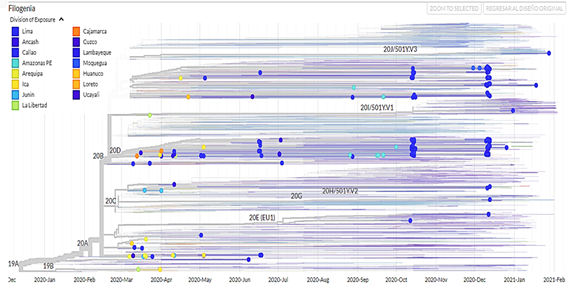
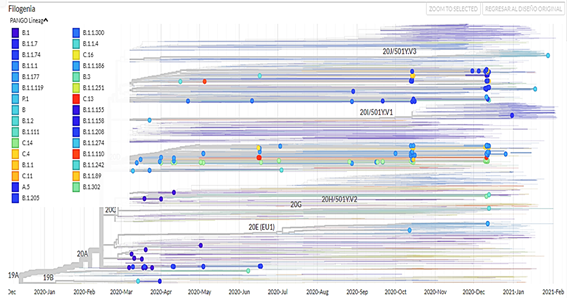
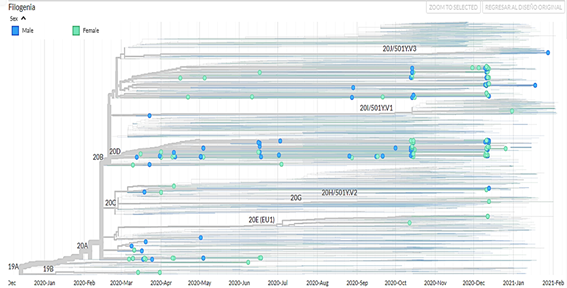
Table 1. Frequency of lineages and clades by gender isolated from patients in Peru from March 2020 to March 2021.
| Lineage | Female | Male | Total | % | p |
|---|---|---|---|---|---|
| A | 2 | 0 | 2 | 0.27 | 0.278 |
| A.1 | 2 | 0 | 2 | 0.27 | |
| A.2 | 5 | 1 | 6 | 0.8 | |
| A.5 | 3 | 1 | 4 | 0.53 | |
| B | 1 | 1 | 2 | 0.27 | |
| B.1 | 24 | 28 | 52 | 6.93 | |
| B.1.1 | 87 | 93 | 180 | 24 | |
| B.1.1.1 | 41 | 49 | 90 | 12 | |
| B.1.1.10 | 0 | 2 | 2 | 0.27 | |
| B.1.1.110 | 5 | 7 | 12 | 1.6 | |
| B.1.1.158 | 2 | 3 | 5 | 0.67 | |
| B.1.1.16 | 0 | 1 | 1 | 0.13 | |
| B.1.1.166 | 1 | 0 | 1 | 0.13 | |
| B.1.1.207 | 1 | 0 | 1 | 0.13 | |
| B.1.1.220 | 1 | 3 | 4 | 0.53 | |
| B.1.1.221 | 5 | 4 | 9 | 1.2 | |
| B.1.1.25 | 1 | 0 | 1 | 0.13 | |
| B.1.1.274 | 0 | 1 | 1 | 0.13 | |
| B.1.1.28 | 1 | 0 | 1 | 0.13 | |
| B.1.1.348 | 14 | 13 | 27 | 3.6 | |
| B.1.1.370 | 0 | 1 | 1 | 0.13 | |
| B.1.1.372 | 0 | 1 | 1 | 0.13 | |
| B.1.1.381 | 6 | 6 | 12 | 1.6 | |
| B.1.1.485 | 4 | 6 | 10 | 1.33 | |
| B.1.1.500 | 1 | 1 | 2 | 0.27 | |
| B.1.1.7 | 3 | 0 | 3 | 0.4 | |
| B.1.111 | 1 | 2 | 3 | 0.4 | |
| B.1.13 | 1 | 0 | 1 | 0.13 | |
| B.1.177 | 3 | 1 | 4 | 0.53 | |
| B.1.177.45 | 2 | 0 | 2 | 0.27 | |
| B.1.2 | 4 | 1 | 5 | 0.67 | |
| B.1.205 | 15 | 12 | 27 | 3.6 | |
| B.1.234 | 0 | 2 | 2 | 0.27 | |
| B.1.547 | 0 | 1 | 1 | 0.13 | |
| B.1.596 | 0 | 1 | 1 | 0.13 | |
| B.1.610 | 0 | 4 | 4 | 0.53 | |
| B.1.8 | 0 | 1 | 1 | 0.13 | |
| B.3 | 1 | 0 | 1 | 0.13 | |
| C.11 | 13 | 23 | 36 | 4.8 | |
| C.13 | 7 | 14 | 21 | 2.8 | |
| C.14 | 58 | 52 | 110 | 14.67 | |
| C.22 | 1 | 1 | 2 | 0.27 | |
| C.23 | 0 | 1 | 1 | 0.13 | |
| C.25 | 1 | 6 | 7 | 0.93 | |
| C.32 | 12 | 9 | 21 | 2.8 | |
| C.33 | 5 | 4 | 9 | 1.2 | |
| C.37 | 4 | 3 | 7 | 0.93 | |
| C.4 | 20 | 29 | 49 | 6.53 | |
| N.4 | 0 | 2 | 2 | 0.27 | |
| P.1 | 0 | 1 | 1 | 0.13 | |
| Total | 358 | 392 | 750 | 100 | |
| Clado | Femenino | Masculino | Total | % | p |
| G | 34 | 37 | 71 | 9.47 | 0.0264 |
| GH | 10 | 14 | 24 | 3.2 | |
| GR | 292 | 335 | 627 | 83.6 | |
| GRY | 3 | 0 | 3 | 0.4 | |
| GV | 5 | 1 | 6 | 0.8 | |
| L | 1 | 0 | 1 | 0.13 | |
| O | 1 | 2 | 3 | 0.4 | |
| S | 12 | 2 | 14 | 1.87 | |
| V | 0 | 1 | 1 | 0.13 | |
| Total | 358 | 392 | 750 | 100 |
Table 2. Frequency of lineages and clades by region isolated from patients in Peru from March 2020 to February 2021.
| Lineage | AMA | ANC | APU | ARE | AYA | CAJ | CAL | CUS | HCV | HUA | ICA | JUN | LAL | LAM | LIM | LOR | MOQ | PAS | PIU | SMN | TAC | UCA | Total | p |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | <0.0001 |
| A.1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | |
| A.2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 8 | |
| A.5 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 9 | |
| B | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| B.1 | 0 | 2 | 0 | 8 | 0 | 0 | 5 | 1 | 2 | 0 | 1 | 14 | 0 | 2 | 38 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 74 | |
| B.1.1 | 11 | 4 | 1 | 21 | 1 | 0 | 17 | 1 | 1 | 6 | 9 | 1 | 1 | 3 | 160 | 33 | 2 | 1 | 8 | 4 | 1 | 1 | 287 | |
| B.1.1.1 | 3 | 5 | 0 | 0 | 0 | 0 | 5 | 0 | 0 | 2 | 5 | 1 | 0 | 3 | 85 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 113 | |
| B.1.1.10 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| B.1.1.110 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 12 | |
| B.1.1.158 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | |
| B.1.1.16 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.1.166 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.1.207 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.1.220 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | |
| B.1.1.221 | 0 | 1 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 9 | |
| B.1.1.25 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | |
| B.1.1.274 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.1.28 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| B.1.1.319 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | |
| B.1.1.348 | 0 | 3 | 0 | 2 | 0 | 0 | 3 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 43 | 4 | 2 | 0 | 0 | 0 | 0 | 0 | 60 | |
| B.1.1.370 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.1.372 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| B.1.1.381 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 10 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 13 | |
| B.1.1.398 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.1.434 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.1.485 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 10 | |
| B.1.1.500 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| B.1.1.54 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.1.7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | |
| B.1.111 | 0 | 0 | 0 | 2 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 7 | |
| B.1.13 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.177 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | |
| B.1.177.45 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| B.1.2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | |
| B.1.205 | 0 | 1 | 0 | 1 | 0 | 0 | 4 | 2 | 0 | 0 | 3 | 1 | 0 | 0 | 26 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 38 | |
| B.1.234 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| B.1.243 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.298 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.547 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| B.1.557 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.561 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | |
| B.1.596 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.1.610 | 0 | 0 | 0 | 2 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 12 | |
| B.1.8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| B.3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| C.11 | 0 | 6 | 0 | 1 | 0 | 1 | 4 | 0 | 4 | 11 | 3 | 5 | 1 | 3 | 53 | 9 | 0 | 0 | 1 | 0 | 0 | 0 | 102 | |
| C.13 | 0 | 1 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 1 | 2 | 0 | 0 | 1 | 19 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 28 | |
| C.14 | 13 | 22 | 0 | 0 | 3 | 0 | 12 | 1 | 4 | 2 | 2 | 2 | 1 | 25 | 70 | 3 | 0 | 0 | 3 | 1 | 0 | 0 | 164 | |
| C.22 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | |
| C.23 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| C.25 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 8 | |
| C.32 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 31 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 33 | |
| C.33 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 10 | |
| C.37 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 48 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 51 | |
| C.4 | 1 | 0 | 0 | 0 | 0 | 0 | 7 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 57 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 67 | |
| C.8 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | |
| N.4 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| P.1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 3 | 16 | 0 | 0 | 0 | 0 | 0 | 0 | 24 | |
| Total | 31 | 49 | 3 | 42 | 5 | 3 | 77 | 7 | 11 | 38 | 28 | 27 | 5 | 40 | 750 | 74 | 4 | 1 | 18 | 5 | 1 | 1 | 1220 | |
| Clado | AMA | ANC | APU | ARE | AYA | CAJ | CAL | CUS | HCV | HUA | ICA | JUN | LAL | LAM | LIM | LOR | MOQ | PAS | PIU | SMN | TAC | UCA | Total | p |
| G | 0 | 1 | 0 | 11 | 0 | 0 | 9 | 3 | 2 | 2 | 4 | 6 | 0 | 1 | 67 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 108 | <0.0001 |
| GH | 0 | 2 | 0 | 2 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 9 | 0 | 1 | 22 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 39 | |
| GR | 31 | 45 | 3 | 29 | 5 | 2 | 63 | 4 | 9 | 36 | 22 | 12 | 3 | 36 | 632 | 72 | 4 | 1 | 18 | 5 | 1 | 1 | 1034 | |
| GRY | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | |
| GV | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | |
| L | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| O | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | |
| S | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 2 | 0 | 2 | 0 | 16 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 22 | |
| V | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
AMA: Amazonas; ANC: Ancash; APU: Apurímac; ARE: Arequipa, AYA: Ayacucho; CAJ: Cajamarca; CAL: Callao; CUS: Cusco; HCV: Huancavelica; HUA: Huánuco; ICA: Ica; JUN: Junín; LAL: La Libertad; LAM: Lambayeque; LIM: Lima; LOR: Loreto; MOQ: Moquegua; PAS: Pasco; PIU: Piura; SMN: San Martín; TAC: Tacna; UCA: Ucayali.
DISCUSSION
The analysis of the different genomes has been very useful to track the SARS-CoV-2 virus in different regions, not only in Peru but also in Latin America and the world. Regarding clades, Peru has identified the GR clade as in all South American countries with the exception of Paraguay, which has not identified SARS-CoV-2 genomes to date; on the other hand, clade G is common among all countries except Venezuela and clade GH in all except Bolivia and Uruguay, these being the most common, followed by clades S, L and GV. Other clades identified in this continent but not in Peru were clades O and V11.
In Uruguay, genetic analysis shows that clades S and G (G, GH, GR) dominate, representing more than 90% of the virus strains in our study; the fatal outcome of SARS-CoV-2 infection is significantly related to hypertension, renal failure and ICU admission (FDR <0.01), but there are no mutations in structural or non-structural proteins such as the D614G peak protein12. As in Peru, North American countries such as Canada have tracked the GR clade more frequently, followed by the G and GH clades and the S and L clades less regularly. The latter were identified at the same frequency in the United States. However, in this country the clade GH has been identified with more frequency followed by the clades GR and G; In Mexico, the same does not happen, since the most frequent clades are GR and G followed by GH, being the same in the case of clades S and L being very minor, in addition to the fact that in this part of the American continent clades were found O and V11. In Europe and Asia, as in Peru, the clades GR, G, and GH have been registered more, and below are the clades S, L, and GV, taking into account that in these continents, a greater number of analyzes of genomes of the virus have been carried out SARS-CoV-211.
Regarding the viral lineages, differences were found with the genomes isolated in Argentina since the lineage found with the highest frequency and persistence was B.1 followed by B.1.1.33 and B.1.499; the same happens in Bolivia. Although only 14 genomes have been identified, the B.1 lineage persisted through time; in Brazil and Uruguay, the lineages B.1.1.33 and B.1.1.28 were present for approximately the entire time spanned by the pandemic in that country. While in Chile, Colombia and Ecuador, the most frequent and persistent lineages are B.1 and B.1.1.74. In North America, Europe and Asia, the majority lineage is B.1 but in the middle and end of 2020, there was an increase in lineages B.1.2, B.1.1.7 and B.1.17713, data that differ with the present study.
In addition to the analysis of genomes found on web platforms, studies published in journals should be considered, as is the case of the research carried out in Ecuador where the most dominant lineage in all sequences from that country was B.1.1.74, followed by B.1 and B.1.1.114, which does not coincide with our results. On the other hand, the P.1 lineage was identified in cities of the Brazilian Amazon such as Manaus15, which is related to its transmission in the Loreto region where it was the most frequent lineage after B.1.1. Regarding a study carried out in Venezuela, the lineages B.1.1 and B.1.111 were the most persistent in different provinces16, which is not related to the present investigation, like the lineages identified with the highest incidence in South Africa as are C.1, B.1.1.56 and B.1.1.5417.
In the case of lineages by regions, Lima has presented the highest number of identified lineages and clades. Therefore, it is suggested that it presents the highest number of infections in that city followed by Callao, Loreto, Áncash, and Arequipa, making the transmissibility of the virus in those cities is very high.
It should be noted that the B.1.1.7 lineage has been very important in terms of the hospitalization of patients in a serious way or in their death, as indicated by a study from the United Kingdom, where more than 60% of patients sampled presented these characteristics18, as in the United States where the rapid transmission of this lineage caused an increase in deaths19, which would be related to the present study, since the decreased manifestation of this lineage in Peru did not it is expressed in a disproportionate increase in hospitalizations and deaths, as occurred in the European country. Finally, in a study carried out in India, it has been shown that the clades GR, GH, and G are the most predominant with more than 70%20, coinciding with our results.
This retrospective study has limitations such as the small sample size, there is no control over the quality of the data that is uploaded, and the bias in data collection.
For these reasons, it is important to carry out a genomic epidemiological surveillance since it could give us a glimpse of the new changes in the variants of SARS-CoV-2 and the use of this information to take containment measures, such as new forms of diagnosis, monitoring, clinic, etc. It is important to track how it spreads and whether the mutations are clinically important in infected patients. This is achieved in a joint work of the state with the researchers.
CONCLUSION
The clade GR is common to all South American countries, European and Asian continents, followed by clades G and GH most frequently; on the other hand, the most persistent viral lineage in Peru is B.1.1, this being not common with other countries. In addition, most of the lineages were identified in the Lima, Callao, Loreto, Ancash, and Arequipa regions.
Acknowledgments:
To the National Institute of Health of Peru, Universidad Peruana Cayetano Heredia, National Center for Epidemiology, Prevention and Control of Diseases - CDC PERU, Hospital Regional Lambayeque for having your data free to carry out this study
REFERENCES
1. Iglesias-Osores S. Contagio y medidas de prevención de SARS-CoV-2 (COVID-19) en prisiones. Rev Española Sanid Penit. 2020 Jun;22(2):92-96. [ Links ]
2. Rodriguez-Morales AJ, Cardona-Ospina JA, Gutiérrez-Ocampo E, Villamizar-Peña R, Holguin-Rivera Y, Escalera-Antezana JP, et al. Clinical, laboratory and imaging features of COVID-19: A systematic review and meta-analysis. Travel Medicine and Infectious Disease. 2020; 34:101623. DOI: 10.1016/j.tmaid.2020.101623. [ Links ]
3. Saavedra-Camacho JL, Iglesias-Osores S. Carrera contra el tiempo: creación de una vacuna contra el COVID-19. Univ Médica Pinareña. 2020; 16(3):3-5. [ Links ]
4. Al-Omari A, Rabaan AA, Salih S, Al-Tawfiq JA, Memish ZA. MERS coronavirus outbreak: Implications for emerging viral infections. Diagn Microbiol Infect Dis [Internet]. 2019;93(3):265-285. Available from: https://sci-hub.se/10.1016/j.diagmicrobio.2018.10.011 [ Links ]
5. Rabaan AA. Middle East respiratory syndrome coronavirus: five years later. Expert Rev Respir Med [Internet]. 2017;11(11):901-912. Available from: https://doi.org/10.1080/17476348.2017.1367288 [ Links ]
6. Kress WJ, Erickson DL. DNA barcodes: Genes, genomics, and bioinformatics. Vol. 105, Proceedings of the National Academy of Sciences of the United States of America. National Academy of Sciences. 2008; 105(8): 2761-2762. DOI: 10.1073/pnas.0800476105. [ Links ]
7. Forster P, Forster L, Renfrew C, Forster M. Phylogenetic network analysis of SARS-CoV-2 genomes. Proc Natl Acad Sci. 2020;117(17): 9241-9243. DOI: 10.1073/pnas.2004999117. [ Links ]
8. Tillett RL, Sevinsky JR, Hartley PD, Kerwin H, Crawford N, Gorzalski A, et al. Genomic evidence for reinfection with SARS-CoV-2: a case study. Lancet Infect Dis. 2021;21(1):52-8. DOI: 10.1016/S1473-3099(20)30764-7. [ Links ]
9. Khailany RA, Safdar M, Ozaslan M. Genomic characterization of a novel SARS-CoV-2. Gene Reports. 2020;19:100682. DOI: 10.1016/j.genrep.2020.100682. [ Links ]
10. Laamarti M, Alouane T, Kartti S, Chemao-Elfihri MW, Hakmi M, Essabbar A, et al. Large scale genomic analysis of 3067 SARS-CoV-2 genomes reveals a clonal geo-distribution and a rich genetic variations of hotspots mutations. Gao F, editor. PLoS One. 2020;15(11):e0240345. DOI: 10.1371/journal.pone.0240345. [ Links ]
11. Nextstrain. Genomic epidemiology of novel coronavirus - Global subsampling [Internet]. Nextstrain. 2021 [cited 2021 Feb 12]. Available from: https://nextstrain.org/ncov/south-america?c=GISAID_clade&f_country=Argentina&lang=es&p=full&tl=originating_lab. [ Links ]
12. Elizondo V, Harkins GW, Mabvakure B, Smidt S, Zappile P, Marier C, et al. SARS-CoV-2 genomic characterization and clinical manifestation of the COVID-19 outbreak in Uruguay. Emerg Microbes Infect. 2021;10(1):51-65. DOI: 10.1080/22221751.2020.1863747. [ Links ]
13. Vega-fernández J, Iglesias-osores S, Tullume-vergara P. Use of a bioinformatic tool for the molecular epidemiology of SARS-CoV-2. Univ Médica Pinareña. 2020;16(3):3-5. Disponible en: http://www.revgaleno.sld.cu/index.php/ump/article/view/530. [ Links ]
14. Gutierrez B, Márquez S, Prado-Vivar B, Becerra-Wong M, Guadalupe JJ, da Silva Candido D, et al. Genomic epidemiology of SARS-CoV-2 transmission lineages in Ecuador. medRxiv. [prepint] 2021. Disponible en: https://www.medrxiv.org/content/10.1101/2021.03.31.21254685v1.full. [ Links ]
15. Faria NR, Mellan TA, Whittaker C, Claro IM, da Silva Candido D, Mishra S, et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science. [Prepint]. 2021. DOI: 10.1101/2021.02.26.21252554 [ Links ]
16. Loureiro CL, Jaspe RC, D'Angelo P, Zambrano JL, Rodriguez L, Alarcon V, et al. SARS-CoV-2 genetic diversity in Venezuela: Predominance of D614G variants and analysis of one outbreak. PLoS One. 2021;16(2): e0247196. DOI: 10.1371/journal.pone.0247196 [ Links ]
17. Tegally H, Wilkinson E, Lessells RJ, Giandhari J, Pillay S, Msomi N, et al. Sixteen novel lineages of SARS-CoV-2 in South Africa. Nat Med [Internet]. 2021;27(3):440-446. Available from: http://dx.doi.org/10.1038/s41591-021-01255-3 [ Links ]
18. Frampton D, Rampling T, Cross A, Bailey H, Heaney J, Byott M, et al. Genomic characteristics and clinical effect of the emergent SARS-CoV-2 B.1.1.7 lineage in London, UK: a whole-genome sequencing and hospital-based cohort study. Lancet Infect Dis [Internet]. 2021;3099(21): 00170-5. Available from: http://dx.doi.org/10.1016/S1473-3099(21)00170-5 [ Links ]
19. Washington NL, Gangavarapu K, Zeller M, Bolze A, Cirulli ET, Schiabor Barrett KM, et al. Emergence and rapid transmission of SARS-CoV-2 B.1.1.7 in the United States. Cell. 2021;1-19. Disponible en: https://doi.org/10.1016/j.cell.2021.03.052. [ Links ]
20. Potdar V, Vipat V, Ramdasi A, Jadhav S, Pawar-Patil J, Walimbe A. Phylogenetic classification of the whole-genome sequences of SARS-CoV-2 from India & evolutionary trends. Indian J Med Res. 2018;153(1):166-174. Disponible en: https://www.ijmr.org.in/article.asp?issn=0971-5916;year=2021;volume=153;issue=1;spage=166;epage=174;aulast=Potdar. [ Links ]
Received: January 25, 2021; Accepted: May 20, 2021
 Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
