










Services on Demand
Journal

Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de la Facultad de Medicina Humana
Print version ISSN 1814-5469On-line version ISSN 2308-0531
Rev. Fac. Med. Hum. vol.22 no.2 Lima Apr./Jun. 2022 Epub Mar 16, 2022
http://dx.doi.org/10.25176/rfmh.v22i2.3816
Clinical case
Congenital Mesoblastic Nephroma: Case Report
1Unidad de Cirugía Pediátrica y Neonatal del Instituto Nacional de Salud San Borja Lima, Perú.
2Unidad de Cirugía Pediátrica y Neonatal del Instituto Nacional de Salud San Borja. Lima, Perú.
3Instituto Nacional de Salud San Borja. Lima, Perú.
Congenital mesoblastic nephroma (CMN) is the most frequent renal tumor in newborns and infants. under 3 months. The clinical case of a patient younger than 3 months, with prenatal diagnosis, referred in a timely manner for evaluation and management by the Institution is presented. CMN is a low incidence tumor. Early diagnosis as well as complete excision of the tumor are predictors of good prognosis, as in the case of our patient.
Keywords: Congenital mesoblastic nephroma; Renal tumor; Infants. (Source : MeSH - NLM).
INTRODUCTION
Congenital mesoblastic nephroma (CMN), also called fetal renal hamartoma, leiomyomatous or mesenchymal hamartoma, is a tumor with a good prognosis, except for the cell line1. Described for the first time by Bolande in 1967, as a tumor different from nephroblastoma due to its histology, treatment and prognosis2. It represents 3 to 5% of all renal tumors in pediatrics3, more frequently in newborns and infants under 3 months2. Local recurrence and metastasis is 5% in the first year4.
The origin is probably due to nephrogenic mesenchymal proliferation5. The NMC has a survival of 85% at 5 years. Metastasis is more frequently local, followed by lung, liver, brain, and heart involvement. Survival after recurrence or with metastasis is 57% at 5 years6.
CLINICAL CASE
It’s the clinical case of a 54-day-old infant, born at term, with no history, from Ancash. Referred to institution with diagnosis of right renal mass on prenatal ultrasound during third trimester. The mother is a young woman with no significant history or harmful habits.
In the physical evaluation of the patient, a mass was palpated on the right flank, neither mobile nor painful. The laboratories did not show alterations, nor did he present any other symptoms. The ultrasound confirmed a heterogeneous tumor with regular borders in the upper 1/3 of the right kidney. (Figura 1a)
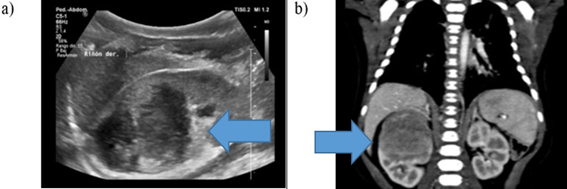
Figura 1: Imaging study: a) Renal ultrasound: heterogeneous tumor with regular borders in the upper 1/3 of the right kidney. b)Abdominal CT with contrast: expansive lesion of slightly heterogeneous density, 41mm x 35mm x 36mm
A computed tomography scan was performed showing an expansive lesion of slightly heterogeneous density, located in the upper half of the right kidney, measuring 41mm x 35mm x 36mm (DL x DBH x SD). The mass was confined to the renal parenchyma, reaching the renal pelvis with mild pyelic ectasia and scant vascularity without significant contrast uptake or internal calcifications in the tumor. The right kidney presents an anatomical variant with two right renal veins. (Figura 1b)
She presented herself to the Institution's Oncology Committee and it was decided to perform a right radical nephrectomy, extracting the piece without complications, and she was discharged 9 days after surgery. The right kidney, right suprarenal gland and right ureter are received in the pathological anatomy department. The tumor measures 4.2cm x 3.9cm x 3cm, located in the upper 2/3 of the right kidney, gray-white in color, fibrous and myxoid in appearance, without necrosis or hemorrhage, representing 50% of the right renal volume.
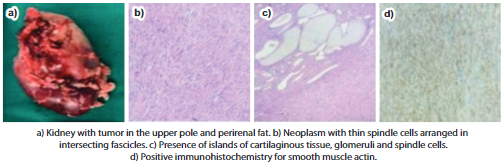
Microscopy revealed CMN, a classic variant, with infiltration of the renal capsule, perirenal fat, renal pelvis, and gerota's fascia. The renal vein, renal sinus, and surgical border of the ureter were free of neoplasia. Immunohistochemistry reports negative WT-1 and Desmin, positive actin and vimentin, and Ki-67 of 2%. (Figure 2)
The patient did not receive neoadjuvant or adjuvant chemotherapy and is controlled in an outpatient clinic, without presenting surgical complications or recurrence, with survival to the date of more than 3 years.
DISCUSSION
Approximately 5% of perinatal tumors arise from the kidney. CMN is the second most common tumor after Wilms' tumor during the first year and is the most common in the first 6 months of life,2as was the case of the patient with 2 months of life. Unlike those published by Geramizadeh, with patients aged between 18 months and 11 years7.
There are 3 histological types: classic (24%), cellular (66%), and mixed (10%) being the classic one with the best prognosis4. Intrauterine manifests with polyhydramnios and hydrops fetalis, associated with premature delivery and hypercalcemia5. The clinical picture presents as a palpable mass in the flank (31.8%), hematuria (27.3%), lumbar pain (22.7%), and hypertension due to increased renin8, it may present pulmonary hypertension and heart failure9.
It is more frequent in females8, differing from the results found by Pachls, where males predominated10. The differential diagnosis is with Wilms tumor, metanephric stromal tumor, and clear cell sarcoma of the kidney, with a worse prognosis1. Obstetric ultrasound allows intrauterine diagnosis2. While the computerized axial tomography of the abdomen allows to make the diagnosis, as well as to differentiate between the histological types5.
The initial treatment is surgical, except for those patients with a high suspicion of cellular histology and older than 3 months, where chemotherapy is indicated from the preoperative period4. Also, when the tumors are larger, chemotherapy allows a reduction, with less risk of intraoperative complications2. Adjuvant chemotherapy is indicated in patients with incomplete tumor resection or tumor rupture during excision, as well as in local recurrence and metastasis.
There are 3 histological variants of CMN: classic, cellular, and mixed. The classic type is characterized by being a solid and firm tumor with the presence of a capsule and fibroblastic cells, with low mitotic activity and abundant collagen deposition. The cell type with long hemorrhagic areas with a necrotic component, high mitotic activity, invasion of peripheral fat and connective tissue11. In immunohistochemistry, CMN is positive for vimentin and smooth tissue actin, negative for desmin and CD3412.
The pathological anatomy of the patient-reported classic-type histology with tumor-free surgical margins. Currently, the patient is free of disease, without recurrence 3 years after surgery. Unlike what was published by Jehangir, with a recurrence of up to 71% and metastasis of 42% in patients with cell-type histology, this being a risk factor13.
Reported poor prognostic factors are age over 3 months, positive surgical margins, cellular histological variant, and tumor rupture during excision14. The patient did not present any of these factors.
CONCLUSIONS
CMN is a low-incidence tumor, more frequent in newborns and infants under 3 months. Good prognostic factors allow greater survival. The initial treatment is surgical, except for patients with risk factors.
Comprehensive evaluation, identifying warning signs and early intervention are factors that will favor survival in pediatric patients with solid abdominal tumors, which is why it is important to know this pathology.
REFERENCES
1. Navarrete R, Leiva J, Castro S, Gilbert C, Ramirez J.Nefroma mesoblastico congenito: A propOsito de un caso. CIRUPED. 2015;5(3):53-7. https://www.researchgate.net/profile/Jorge-Isaac-Ramirez-Rivera/publication/306315811_Nefroma_mesoblastico_congenito_A_proposito_de_un_caso/links/57b7a0d108aec9984ff2b30b/Nefroma-mesoblastico-congenito-A-proposito-de-un-caso.pdf [ Links ]
2. Traore F, Maiga B, Diabate K, Coulibaly Y, Diall H, Togo P, et al. Treatment of congenital mesoblastic nephroma at pediatric oncology unit of GabrielToure teaching hospital. J Pediatr Pediatr Med. 2018;2(3):4-7. DOI: 10.29245/2578-2940/2018/3.1128 [ Links ]
3. El Demellawy D, Cundiff CA, Nasr A, Ozolek JA, Elawabdeh N, Caltharp SA, et al. Congenital mesoblastic nephroma: a study of 19 cases using immunohistochemistry and ETV6-NTRK3 fusion gene rearrangement. Pathology (Phila).2016;48(1):47-50. DOI: https://doi.org/10.1016/j.pathol.2015.11.007 [ Links ]
4. Wang Z-P, Li K, Dong K-R, Xiao X-M, Zheng S. Congenital mesoblastic nephroma: Clinical analysis of eight cases and a review of the literature. Oncol Lett. 2014;8(5):2007-11.DOI: 10.3892/ol.2014.2489 [ Links ]
5. Santos LG dos, Carvalho J de SR de, Reis MA, Sales RUB. Nefroma mesoblastico congenito subtipo celular: relato de caso. J Bras Nefrol. 2011;33(1):109-12. DOI: 10.1590/S0101-28002011000100014 [ Links ]
6. Zaidi Z, Mouriquand P. Congenital Mesoblastic Nephroma. JPMA[Internet]. 1997 [citado el 24 de febrero de 2022];47(9). Disponible en: https://jpma.org.pk/article-details/4209 [ Links ]
7. Geramizadeh B, Kashkooe A, Keshavarz P, Zareifar S, Foroutan H.Cellular mesoblastic nephroma in infants and children: Report of four cases and review of the literature. Urol J. 2020;87(2):91-6. DOI: 10.1177/0391560319850436 [ Links ]
8. Daniel J, Ruzic A, Dalland J, Miller V, Hanna M. Management of mixed type congenital mesoblastic nephroma: Case series and review of the literature. J Neonatal-Perinat Med. 2017;1 0 (1):1 13-8. DOI: 10.3233/NPM-1617 [ Links ]
9. Arias Santos MD, Pavcovich Ruiz M, Andujar Sanchez M, Martinez Lanao D, Le6n Arencibia L. Nefroma mesoblastico congenito. Rev Esp Patol. 2006;39(4):243-5. DOI: 10.1016/S1699-8855(06)70047-2 [ Links ]
10. Pachl M, Arul G, Jester I, Bowen C, Hobin D, Morland B. Congenital mesoblastic nephroma: a single-centre series. Ann R Coll Surg Engl. 2020;102(1):67-70. DOI: 10.1308/rcsann.2019.0111 [ Links ]
11. Tripathy PK, Behera 5, Mohanty HK. Cellular Congenital Mesoblastic Nephroma in a Newborn. J Neonatal Surg. 2017;6(2):45. DOI: 10.21699/jns.v6i2.564 [ Links ]
12. Leao SC, Fernandes DM, Dias BG, Oliveira WR, Oliveira SM de, Rangel MRU. Mixed subtype of congenital mesoblastic nephroma with poor evolution: a case report and literature review. Radio! Bras. 2015;48(6):396-8. DOI: 10.1590/0100-3984.2013.1613 [ Links ]
13. Jehangir 5, Kurian JJ, Selvarajah D, Thomas RJ, Holland AJA. Recurrent and metastatic congenital mesoblastic nephroma: where does the evidence stand? Pediatr Surg Int. 2017;33(1 1):1 183-8. DOI: 10.1007/s00383-017-4149-5 [ Links ]
14. Furtwaengler R, Reinhard H, Leuschner I, SchenkJP, Goebel U, Claviez A, et al. Mesoblastic nephroma-A report from the Gesellschaft fur Pediatrische Onkologie and Hamatologie (GPOH). Cancer. 2006;106(10):2275-83. DOI: 10.1002/cncr.21836 [ Links ]
Received: May 01, 2021; Accepted: February 16, 2022
 Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
