










Servicios Personalizados
Revista

Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Facultad de Medicina Humana
versión impresa ISSN 1814-5469versión On-line ISSN 2308-0531
Rev. Fac. Med. Hum. vol.22 no.4 Lima oct./dic. 2022 Epub 12-Oct-2022
http://dx.doi.org/10.25176/rfmh.v22i4.5098
Review article
Genetic studies to determine the risk of non-ischemic sudden death: review article
1Sequence Reference Lab. Lima, Perú.
2Hospital Guillermo Almenara Irigoyen. Lima, Perú.
3Instituto de investigaciones en ciencias biomédicas. Facultad de medicina humana. Universidad Ricardo Palma. Lima, Perú.
Heart disease is the cause of sudden death in more than 80% of cases. Ischemic heart disease is the cause for 90% of all sudden cardiac deaths, while in the remaining 10% of cases, heart diseases have a hereditary origin and comprise a wide spectrum of disorders that include cardiomyopathies and channelopathies. The aim of this review is to highlight the importance of genetic counseling for patients with hereditary cardiovascular disease and its evaluation by a multidisciplinary team.
Keywords: Heart diseases; Sudden death; Genetics; Genetic Counseling. (fuente: MeSH NLM).
INTRODUCTION
Sudden, unexplained death is considered due to a primary arrhythmic disorder when no structural heart disease is found at autopsy and there is no prior documentation of heart disease.1,2. In the general population, the incidence of sudden death varies from 1/100,000 in adolescents to 1/1,000 in individuals aged 45 to 75 years.3Is believed that channelopathies are responsible for 10% to 15% of cases of sudden unexplained death in young adults and children.4
Hereditary cardiomyopathies are a genetically and phenotypically heterogeneous group that predispose to heart failure and sudden death, due to pathogenic variants in genes that encode essential proteins in the structural components of cardiomyocytes and are functionally and morphologically classified by their characteristics in: hypertrophic cardiomyopathy, cardiomyopathy dilated cardiomyopathy/arrhythmogenic right ventricular dysplasia, cardiomyopathy due to noncompaction of the ventricle, and restrictive cardiomyopathy.5
Channelopathies are a group of hereditary syndromes of cardiac arrhythmia, with variable clinical expression, including Brugada syndrome, catecholaminergic polymorphic ventricular tachycardia, long QT syndrome, short QT syndrome, and early repolarization syndrome.6These channelopathies arise from defects in macromolecular complexes of ion channels or proteins critical for the intracellular handling of calcium, sodium, and/or potassium. Patients with channelopathy usually have a structurally normal heart but are predisposed to syncope/arrhythmic seizures and sudden cardiac death.7
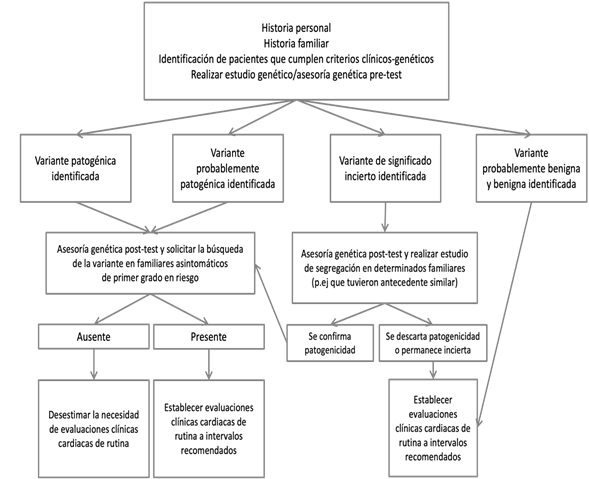
From the discovery of genes associated with hereditary cardiovascular disease of monogenic origin in the early 1990s to the present day, where studies of genetic panels or complete exomes6are carried out in patients with suspected hereditary heart disease, genetic studies meet an important role in supporting the diagnosis of a primary arrhythmia disorder. In addition, they provide prognostic information and allow the evaluation to be extended to asymptomatic first-degree relatives at risk. Genetic studies are used in clinical practice at the time of risk stratification and patient management, based on precision medicine, focused on genomic medicine (Figure 1)8
The aim of this review is to highlight the importance of genetic counseling for patients with hereditary cardiovascular disease and its evaluation by a multidisciplinary team.
Hypertrophic cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is characterized by left ventricular hypertrophy in the absence of an underlying systemic condition or other cardiac disease, such as valvular heart disease or hypertension. It is estimated to have a prevalence of 1 in 500 in the general population.9HCM is the most common inherited type of heart condition and the leading cause of sudden death in young adults and competitive athletes in the United States.10Although the age of onset of HCM can range from infancy to old age, manifestations, in those carrying a pathogenic variant, usually do not appear before adolescence.10HCM is primarily inherited in an autosomal dominant pattern, with reduced penetrance and clinical variability. Clinical manifestations vary from being completely asymptomatic to progressive heart failure and sudden death caused by mechanical or electrical defects.
HCM is defined by the presence of unexplained left ventricular hypertrophy with a maximum wall thickness ≥15 mm in adults or a Z score > 3 in children.11If there is a family history of HCM, or if genetic testing confirms that a family member has inherited any pathogenic variant, LV maximum wall thickness ≥ 13 mm will support the diagnosis. Altered left ventricular relaxation, may be identified in individuals possessing a pathogenic variant in a gene encoding a sarcomere component (nonsyndromic HCM) who have normal left ventricular wall thickness12suggesting that diastolic dysfunction is an early phenotype of HCM rather than a secondary consequence of left ventricular hypertrophy. Although left ventricular hypertrophy and a clinical diagnosis of HCM are often evident during adolescence, around the onset of puberty, or during adulthood, the onset may be earlier (in infancy and childhood).13
HCM is frequently described as a disease of the sarcomere, and pathogenic variants have been detected in almost all sarcomeric proteins.14Molecular mechanisms include increased actin-activated ATPase activity, disruption of actin-myosin interaction, and altered intracellular calcium signaling in cardiomyocytes.15Furthermore, some data suggest that left ventricular hypertrophy may be caused by alterations in the transforming growth factor B and CaMKII Mef2 signaling pathways.16
According to the genetic etiology of HCM, pathogenic variants have been described in several genes encoding sarcomeric proteins, with the majority (80%) being in the MYH7 and MYBPC3 genes.17(Table 1) Pathogenic variants in genes encoding a sarcomere component are found in approximately 50% - 60% of probands (adults and children) with a family history of HCM, and in approximately 20% -30% of probands without a family history of HCM.18.
Approximately 3% - 5% of affected individuals have biallelic variants in 1 gene or heterozygous variants in more than 1 gene.19Recently, the spectrum of genes associated with HCM has expanded to non-sarcomeric genes and includes genes encoding Z-disk proteins and proteins located in the sarcoplasmic reticulum and plasma membrane. However, variants in these genes are rare.
Variants in the MYH7 gene generally lead to significant left ventricular hypertrophy in the second decade of life and are believed to be associated with an increased risk of heart failure and sudden cardiac death.20In contrast, pathogenic variants in MYBPC3 are thought to be associated with older age at diagnosis(18)although they have also been identified in a significant proportion of patients with childhood-onset left ventricular hypertrophy.19
The differential diagnosis of HCM includes various syndromes that usually manifest with multiorgan involvement but may also present with isolated or predominant left ventricular hypertrophy. These syndromes include metabolic depot cardiomyopathies such as Danon disease (LAMP2 gene), Wolff-Parkinson-White syndrome (PRKAG2 gene)21, Fabry disease which is a lysosomal storage disorder (GLA gene)22as well as syndromic (with other systemic involvement) (Table 2) and non-syndromic (without other systemic involvement) disorders.
Table 1. Genes associated with hypertrophic cardiomyopathy.
| Gene | Inheritance | % of HCM caused by pathogenic variant in this gene | Allelic disorders | OMIM (Online Mendelian Inherited in Man) |
|---|---|---|---|---|
| MYBPC3 | AD | 50% | DCM | 600958 |
| MYH7 | AD | 33% | Myosin Deposit Myopathy Cardiomyopathy due to noncompaction of the ventricle | 160760 |
| TNNI3 | AD | 5% | DCM Restrictive cardiomyopathy | 191044 |
| TNNT2 | AD | 4% | DCM Cardiomyopathy due to noncompacted ventricle Familial restrictive cardiomyopathy | 191045 |
| ACTC1 | AD | <3% | DCM | 102540 |
| MYL2 | AD | <3% | 160781 | |
| MYL3 | AD AR | <3% | 160790 | |
| TPM1 | AD | <3% | DCM | 191010 |
| PLN | AD | <3% | DCM | 172405 |
| ALPK3 | AR | Rare | 617608 | |
| ACTN2 | AD | <1% | DCM | 102573 |
| CSRP3 | AD | <1% | DCM | 600824 |
| TNNC1 | AD | <1% | DCM | 191040 |
| JPH2 | AD | Rare | 605267 | |
| MYOZ2 | AD | <1% | 605602 | |
| NEXN | AD | <1% | DCM | 613121 |
| ANKRD1 | AD | Rare | ||
| CALR3 | AD | Rare | 611414 | |
| KLF10 | AD | Rare | 601878 | |
| MYH6 | AD | Rare | DCM | 160710 |
| MYLK2 | Digenic | Rare | 606566 | |
| MYOM1 | AD | Rare | 603508 | |
| MYPN | AD | Rare | DCM Nemaline myopathy | 608517 |
| OBSCN | AD | Rare | 608616 | |
| PDLIM3 | AD | Rare | 605889 | |
| RYR2 | AD | Rare | Arrhythmogenic cardiomyopathy of the right ventricle Catecholaminergic polymorphic ventricular tachycardia | 180902 |
| TCAP | AD | Rare | DCM | 604488 |
| TRIM63 | AD | Rare | 606131 | |
| TTN | AD | Rare | DCM Hereditary myopathy with early respiratory failure Salih distal myopathy Distal myopathy Udd | 613765 |
| VCL | AD | Rare | DCM | 193065 |
AD: autosomal dominant; AR: Aautosomal recessive; DCM: Dilated cardiomyopathy.
Table 2. Syndromic hypertrophic cardiomyopathy.
| Disorder | Gene(s) | Heritage | Other clinical features (non-cardiac) |
|---|---|---|---|
| Danon’s disease | LAMP2 | XL | ● Myopathy ● Retinal dystrophy |
| Fabry disease | GLA | XL | ● Periodic pain crises in the extremities ● Angiokeratomas ● Hypohidrosis ● Ocular abnormalities ● Proteinuria and deterioration of renal function |
| Friedreich’s ataxia | FXN | AR | ● Ataxia, slowly progressive before 25 years of age ● Dysarthria ● Muscle weakness |
| Glycogen storage disease of the heart, congenital lethal | PRKAG2 | AD | ● Neonatal hypoglycemia ● Vacuolar myopathy ● Facial dysmorphia and/or macroglossia |
| Hereditary transthyretin amyloidosis | TTR | AD | ● Slow progressive peripheral sensory-motor neuropathy and autonomic neuropathy ● Vitreous opacity ● CNS amyloidosis |
| Pompe disease | GAA | AR | ● Poor nutrition ● Macroglossia ● Motor delay / muscle weakness ● Respiratory distress |
| Rasopathies: ● Noonan syndrome ● Cardiofasciocutaneous syndrome ● Costello syndrome ● Noonan syndrome with multiple lentigines | BRAF HRAS KRAS LZTR1 MAP2K1 MAP2K2 NRAS PTPN11 RAF1 RASA2 RRAS RIT1 SOS1 SOS2 | AD | ● Facial characteristics ● Short stature ● Variable developmental delay ● Broad, winged neck |
Dilated cardiomyopathy
Dilated cardiomyopathy (DCM) is defined by left ventricular dilatation and systolic dysfunction, and is the most common indication for cardiac transplantation in the United States.23The spectrum of clinical manifestations includes heart failure, thromboembolism, and sudden death. The estimated prevalence of idiopathic type DCM is 1 in 2500 individuals. The percentage of idiopathic CMD that have a genetic etiology is estimated to be between 30% and 50% depending on the presence of family history.23The age of onset can range from childhood to late adulthood, although most patients are diagnosed between 20 and 50 years of age.24Compared to HCM, which is primarily a sarcomere disease, DCM presents greater genetic heterogeneity (more than 40 genes described to date).25
DCM is mainly inherited in an autosomal dominant pattern, however, X-linked inheritance has also been described.26Due to the locus and allelic heterogeneity of DCM, gene panels are the most frequently used genetic tests. It is estimated that pathogenic variants can be identified in 17% to 30% of individuals with DCM when multigene panel analysis of up to 20 genes is performed.27Pathogenic variants in the LMNA and SCN5 genes are clearly associated with DCM and conduction system disease.28Pathogenic variants in the TTN gene are identified in up to 25% of familial cases and 18% of sporadic cases of DCM.29(Table 3).
Individuals with CMD in whom the cause has been determined not to be acquired (secondary) or syndromic (with other systemic involvement) (Table 4), have nonsyndromic DCM (without other systemic involvement).
Table 3. Genes associated with dilated cardiomyopathy.
| Gene | Heritage | % of HCM caused by pathogenic variant in this gene | Allelic disorders | OMIM (Online Mendelian Inherited in Man) | Gene |
|---|---|---|---|---|---|
| ACTC1 | <1% | AD | Family HCM | 102540 | |
| ACTN2 | <1% | AD | Family HCM | 102573 | |
| ANKRD1 | 2.2% | AD | 609599 | ||
| BAG3 | 2.5% | AD | Progressive Myofibrillar Myopathy | 603883, PS601419 | |
| CSRP3 | <1% | AD | Family HCM | 600824 | |
| DES | <1% | AD | Arrhythmia and neuromuscular involvement | Myofibrillar Myopathy | 125660, PS601419 |
| DMD | ? | XL | Neuromuscular involvement | Dystrophinopathies | 300377 |
| DSG2 | AD | Possible right ventricular involvement | 125671 | ||
| EYA4 | ? | AD | Deafness | Non-syndromic deafness | 603550 |
| LDB3 | 1% | AD | Myofibrillar myopathy | 605906, PS601419 | |
| LMNA | 6% | AD | Arrhythmia and conduction system disease | Partial lipodystrophy Charcot Marie Tooth 2B1 Emery-Dreifuss muscular dystrophy Hutchinson-Gilford Progeria Syndrome Atypical Werner's syndrome LMNA-related muscular disease | 150330 |
| MYBPC3 | 2%-4% | AD | Family HCM | 600958 | |
| MYH6 | 3%-4% | AD | Family HCM | 160710 | |
| MYH7 | 4.2% | AD | Laing's distal myopathy Family HCM Deposit myopathy (MYH7) Noncompaction ventricular Cardiomyopathy | 160760 | |
| NEXN | <1% | AD | Family HCM | 613121 | |
| PLN | ? | AD | Arrhythmia and conduction system disease | 172405 | |
| PSEN1 | <1% | AD | Early-onset Alzheimer's disease | 104311 | |
| PSEN2 | <1% | AD | Early- and late-onset Alzheimer's disease | 600759 | |
| RBM20 | 1.9% | AD | Arrhythmia and conduction system disease | 613171 | |
| SCN5A | 2%-4% | AD | Arrhythmia and conduction system disease | Long QT syndrome type 3 Brugada Syndrome Idiopathic ventricular fibrillation Sick sinus syndrome Cardiac conduction system disease | 600163 |
| SGCD | <1% | AD | Delta sarcoglycanopathy | 601411 | |
| TAZ | ? | XL | Childhood presentations | Barth's syndrome Endocardial fibroelastosis type 2 Familial noncompaction ventricular cardiomyopathy | 300394 |
| TCAP | 1% | AD | Family HCM | 604488 | |
| TMPO | 1.1% | AD | 188380 | ||
| TNNC1 | <1%-1.3% | AD | Family HCM | 191040 | |
| TNNI3 | 1.3% (AD) <1% (AR) | AD | Family HCM Restrictive cardiomyopathy | 191044 | |
| TNNT2 | 2.9% | AD | Family HCM Noncompaction ventricular cardiomyopathy Family restrictive cardiomyopathy related to TNNT2 | 191045 | |
| TPM1 | <1%-1.9% | AD | Family HCM | 191010 | |
| TTN 4 | 10%-20% | AD | Proximal myopathy, with early respiratory muscle involvement. Tibial muscular dystrophy | 188840 | |
| VCL | 1% | AD | 193065 |
AD: autosomal dominant; AR: Aautosomal recessive; XL: X-linked; mtDNA: Mitochondrial DNA; Mat: Maternal inheritance.
Table 4. Syndromic dilated cardiomyopathy.
| Disorder | Gene(s) | Heritage | Other clinical features (non-cardiac) | Observations |
|---|---|---|---|---|
| Barth syndrome | TAZ | XL | ● Neutropenia ● Muscle weakness ● Delayed development | |
| Carvajal syndrome | DSP | AR | ● Wooly hair ● Palmoplantar keratoderma | |
| Duchenne / Becker muscular dystrophy | DMD | XL | In males: ● Muscle weakness ● Elevated serum CK levels ● Loss of ambulation in childhood | Heterozygous females may present isolated DCM. |
| Emery-Dreifuss muscular dystrophy | EMD FHL1 LMNA | XL AD AR | ● Joint contractures ● Elevated serum CK levels Muscle weakness in childhood/early adulthood | Conduction system diseases and/or arrhythmias are frequent. |
| Hereditary hemochromatosis | HFE | AR | ● Cirrhosis ● Diabetes ● Hypermelanotic pigmentation ● Increased serum iron and ferritin levels. | Non-dilated and/or infiltrative cardiomyopathy is more frequent than DCM. |
| Laing's distal myopathy | MYH7 | AD | ● Facial weakness ● Weakness (childhood onset) of ankles, big toes, finger extensors and neck flexors. | |
| Limb-girdle muscular dystrophy IB | LMNA | AD | Proximal weakness of the lower extremities | Diseases of the conduction system and/or arrhythmias are frequent. |
| Mitochondrial DCM | DNAmt | Mat | Complex phenotypes including: - Focal segmental glomerulosclerosis. - Kearns-Sayre syndrome |
Genetic testing should be offered to all individuals of any age with non-ischemic DCM30, including those with peripartum or pregnancy-associated cardiomyopathy31. The purpose of establishing a molecular diagnosis of DCM is to inform the risk assessment of the patient's relatives.
Pathogenic, probably pathogenic and variants of uncertain significance in more than 30 genes have been reported in 40%-50% of individuals with familial DCM (when there are more than two affected first-degree relatives)32or in isolated cases (in only one family member)33. The detection rate of pathogenic and probably pathogenic variants is approximately 27%34.
Currently, genetic studies through multigene panels have allowed the simultaneous and parallel analysis of all DCM-related genes. Given the complexity of interpreting the results of genetic testing and their implications for follow-up and management, these cases should be discussed in a multidisciplinary manner that includes the corresponding genetic counseling.35
Screening in asymptomatic first-degree relatives of an individual with DCM may allow for early detection, immediate initiation of treatment, and improved long-term outcomes.36Clarification of the genetic status of first-degree family members of an individual with DCM may inform the indication and frequency of appropriate cardiovascular evaluations.
Arrhythmogenic right ventricular cardiomyopathy
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is defined by myocyte loss and fibrofatty infiltration in the myocardium, which is associated with increased susceptibility to arrhythmias and sudden death, and accounts for a significant portion of sudden deaths in athletes and young adults.37ARVC is typically inherited under an autosomal dominant pattern with reduced penetrance and variable expressivity, affecting males more frequently than females.38
In young adults, 80% of cases are diagnosed before the age of 40 years. Definitive diagnosis can be challenging for the clinician due to phenotypic overlap with other genetic and acquired cardiomyopathies.
ARVC is primarily described as a disease of the desmosome, a multiprotein complex that forms cell-to-cell junctions and binds intermediate filaments of adjacent cells, thereby establishing a functional intercellular network. Desmosomes are especially prevalent in tissues that are subjected to mechanical stress, such as cardiac muscle and skin, which explains why the phenotypic spectrum of ARVC encompasses cardiac and cutaneous manifestations. Molecular mechanisms of ARVC include impaired cell-cell adhesion and defective transmission of contractile force.39
Most of the pathogenic variants in ARVC are present in the JUP, DSP, DSC2, DSG2 and PKP2 genes.40In addition, homozygous or compound heterozygous variants in the DSP and JUP genes have been described in patients with DCM or ARVC.41Some non-desmosomal genes have also been described, including the TMEM43 gene.42Analysis of the coding sequence of desmosomal genes can identify a pathogenic variant in up to 50% of individuals with ARVC, with 40% of cases being due to a variant in the PKK2 gene.40
Cardiomyopathy due to non-compaction ventricle
Isolated non-compaction ventricular cardiomyopathy (NVC) is characterized by a highly trabeculated appearance of the LV myocardium. It is believed that the arrest of myocardial compaction during the first trimester of embryonic development is the cause, however, several authors have proposed that it may be an acquired process based on observations of cases of patients who develop NVC after having normal echocardiographic findings. previous.43
The actual prevalence of NVC is unknown, however, there are several reports indicating that it ranges from 0.014% to 1.3%.43Patients with NVC tend to have early-onset disease, with clinical expression ranging from asymptomatic to progressively impaired cardiac function, ventricular hypertrophy, increased thromboembolic events and sudden death.44The left ventricle is usually affected, but 50% of patients have right ventricular involvement in addition.45
The World Health Organization lists NVC as an unclassified cardiomyopathy46as does the European Society of Cardiology47. On the other hand, the American Heart Association classified CNVC as a primary genetic cardiomyopathy in 200648.
Variants have been described in known genes associated with DCM and HCM that encode components of the sarcomere (ACTC1, MYH7, MYBPC3 and TNT2)49, the Z-disc (LDB3)50, the nuclear lamina (LMNA)51, the dystrophin-associated glycoprotein complex (DTNA)52, as well as the Bart syndrome gene (TAZ).53
Restrictive cardiomyopathy
Restrictive cardiomyopathy (RCM) is characterized by increased stiffness of the ventricular chambers, although ventricular wall thickness and systolic function are generally within normal limits. The majority of patients with RCM develop heart failure, leading to early death.54
Several reports suggest that there is a clinical overlap between RCM and HCM.55Recent studies have identified pathogenic variants in sarcomeric protein-coding genes, including TNNI3, TNNT2, MYH7, and ACTC1.56. In addition, nonsense variants in the desmin gene (DES) have been identified in several families with desmin-related myopathy, which may present with CMR.57
The European Heart Rhythm Association Guidelines recommend targeted genetic testing for the specific genetic variant in all at-risk family members following identification of a pathogenic variant in the patient. testing.58
Long QT syndrome (LQTS)
Long QT syndrome is characterized by excessive prolongation of ventricular repolarization associated with an increased risk of ventricular tachyarrhythmias, syncope, or sudden death in patients with a morphologically intact heart. 2,59 The estimated prevalence ranges between 1:2,500 and 1:5,000. However, due to the probability that 2/3 of the patients are underdiagnosed and that approximately between 10% and 35% of them have a normal QTc-corrected interval (QTc), it is possible that the real prevalence is higher.21,22,60. LQTS can be caused by both genetic factors and acquired factors. Congenital LQTS is an inherited disease caused by different mutations in the genes that code for Na+ or K+ transmembrane ion channel proteins.22
There are two types of classic congenital LQTS: Jervell and Lange-Nielson syndrome (long QT syndrome and deafness) and Romano Ward syndrome (isolated QT interval prolongation). Both are related to episodes of sudden cardiac death due to ventricular fibrillation or Torsades de Pointes arrhythmias that ultimately lead to death.61In total, up to 15 types of LQTS have been identified, which cause a slowdown in the inactivation process of the depolarizing current of sodium into the cell and retard the repolarizing current of potassium out of the cell. All of the above causes an increase in depolarization and dispersion of repolarization.62
The mortality rate of untreated patients with LQTS is estimated to be approximately 1% - 2% per year. The incidence of sudden cardiac death varies from family to family depending on the specific genotype. The international LQTS registry reports that the frequency of cardiac events was significantly higher in individuals with LQTS1 (63%) or LQTS2 (46%) compared with patients with LQTS3 (18%).63In addition, the mean range QTc was significantly longer in the LQTS3 group (510 ± 48 ms) compared to the LQTS1 (490 ± 43 ms) or LQTS2 (495 ± 43) groups.64In addition, it was found that although cumulative mortality up to age 40 was similar in the groups of the three genotypes, the probability of dying during a cardiac event was higher in LQTS3 individuals (20%) than in LQTS1 and LQTS2 individuals (4% for each group).64From the genetic point of view, LQTS is a heterogeneous condition, which has an incomplete penetrance (40%).65
Although mutations affecting genes KCNQ1 (LQTS1), KCNH2 (LQTS2), and SCN5A (LQTS3) are responsible for more than 90% of genetically defined cases of LQTS66(Table 5), they have also been described less common forms (<1% of cases) with heterogeneous molecular etiologies (AKAP9, ANK2, CACNA1C, CALM1, CALM2, CAV3, KCNE1, KCNE2, KCNJ2, KCNJ5, SCN4B, SNTA1) ultimately expressed as QT interval prolongation and increased risk of fatal ventricular arrhythmias. There is significant variability in the rates of LQTS events, even within the same genetic subgroup, which leads to a diversity of clinical and genetic variables that confer (according to their combination of presentation in each patient) different types of risk for each patient. individual.67,68
Also, it was determined that the appearance of arrhythmic events during childhood is an important predictor of risk since it has been reported that people who have their first event in this period have a very high risk to present future events.69
LQTS prognosis is also related to the underlying genetic abnormality. Some data from the International LQTS Registry show that the frequency of clinical events, before the start of treatment, from birth to the age of 40 years, was significantly higher in patients with LQTS2 (46%) and LQTS3 (42%) compared with those with LQTS1 (30%).68A higher lethality and lower response to treatment have also been reported for events related to LQTS3 than with other subtypes.69Pathogenic variants that result in amino acid changes in regions Specific ion channel pathways also confer an increased risk of arrhythmia.2
Table 5. Genes associated with long qt syndrome.
| Gene | LQTS phenotype | % of LQTS attributed to pathogenic variants in this gene | Proportion of pathogenic variants detected by given methodology | |
|---|---|---|---|---|
| Sequencing analysis | Deletion/duplication analysis | |||
| KCNH2 | LQTS type 2 | 25%-30% | 97%-98% | 2%-3% |
| KCNQ1 | LQTS type 1 | 30%-35% | 97%-98% | 2%-3% |
| SCN5A | LQTS type 3 | 5%-10% | All variants reported to date | None reported |
The confirmatory diagnosis of LQTS will be made in patients who meet the clinical criteria for disease and the presence of a pathogenic variant in any of the genes involved identified by genetic testing; through, analysis of specific genes, multigene panels and/or more complete exome or genomic tests. The sequencing analysis of a specific gene will be carried out in the event that the familiar pathogenic variant is known.70
Of people who die from complications of LQTS, death is the first sign of the disorder in an estimated 10% to 15%. Long QT syndrome registry studies including patients, individuals with a pathogenic variant (mostly treated), and also relatives who died suddenly show cumulative mortality before the age of 40 years of 6%-8% in type 1 LQTS, type 2, and type 3 phenotypes.71,72In individuals between the ages of 0 and 18 years, those with a type 1, type 2, or type 3 LQTS phenotype had cumulative mortality of 2%, 3%, and 7%, respectively.(71)From ages 19 to 40, mortality rates were 5%, 7%, and 5%, respectively.(71)Although. syncopal events are more common in type 1 LQTS phenotype (63%), followed by type 2 LQTS phenotype (46%) and type 3 LQTS phenotype (18%), the incidence of death is similar in all three.71)
For the type 1 LQTS phenotype (a specific pathogenic variant), a severe increase in mortality was observed during childhood (ages 1-19 years), for the type 2 phenotype, an increase in mortality was observed between ages 15 and 39 years, and in the type 3 phenotype, an increase in mortality was observed between the ages of 15 and 19 years.73
Some types of LQTS are associated with a phenotype that extends beyond cardiac arrhythmia72:
Andersen-Tawil syndrome (LQTS type 7) is associated with a prolonged QT interval, muscle weakness, and facial dysmorphism.
Timothy syndrome (LQTS type 8) is characterized by a prolonged QT interval and neurodevelopmental features of the hands/feet, facial.
Jervell and Lange-Nielson syndrome (JLNS), an LQTS disorder associated with pathogenic biallelic variants KCNQ1 or KCNE1, is associated with profound sensorineural hearing loss.
It is important to note that LQTS associated with biallelic or heterozygous pathogenic variants in two different genes (digenic pathogenic variants) is generally associated with a more severe phenotype with a longer QT interval and a higher incidence of cardiac events.74To date Specific genotype-phenotype correlations have not been established. Additionally, LQTS exhibits reduced penetrance relative to ECG findings. Approximately 25% of people with a pathogenic variant have a normal QT interval (<440 ms) on baseline ECG. In a study conducted by Priori, it was determined that the percentage of genetically affected individuals with the presence of a normal QTc was higher in the LQTS type 1 group (36%) than in the LQTS type 2 (19%) or type 3 (10%) group. %).75In addition, the penetrance of symptoms is also reduced, at least 37% of patients with type 1 LQTS phenotype, 54% with type 2 phenotype and 82% with type 3 phenotype remain asymptomatic.76
Short QT syndrome (SQTS)
Short QT syndrome (SQTS) is an autosomal dominant inherited channelopathy characterized primarily by a shorter-than-normal QT interval on EKG (<350 ms), atrial and ventricular arrhythmias, and a predisposition to sudden cardiac death.77,78Because it is a rare condition, data on its prevalence and demographics are still limited.
Most patients with SQTS have a family history of sudden death and/or AF. The age at the onset of clinical manifestations may be childhood, which is why it has been classified as a possible cause of sudden infant death. The severity of the clinical manifestations of short QT syndrome is highly variable, from asymptomatic to AF, recurrent syncope, and sudden death.79
Genetic analyzes reveal that SQTS is a genetically heterogeneous disease (Table 6) with pathogenic gain-of-function variants in the KCNH2 gene, giving rise to short QT syndrome type 1 (SQTC1), in the KCNQ1 gene associated with short QT syndrome type 2 (SQTS2), in the KCNJ2 gene associated with short QT syndrome type 3 (SQTS3) and loss-of-function variants with a Brugada syndrome phenotype in the CACNA1C gene, giving rise to short QT syndrome type 4 (SQTS4) and in the CACNB2 gene, giving rise to short QT syndrome type 5 (SQTS5).80Data from a cohort of patients suggest that carriers of KCNH2 mutations may have a shorter QT interval.81
Table 6. Genes associated with short qt syndrome.
| Subtype | Mode of inheritance | Gene | Net effect of mutation |
|---|---|---|---|
| SQTS1 | AD | KCNH2 | IKr function gain |
| SQTS2 | AD | KCNQ1 | Function gain of IKs |
| SQTS3 | AD | KCNJ2 | Function gain of IK1 |
| SQTS4 | AD | CACNA1C | Loss of function of ICa. |
| SQTS5 | AD | CACNB2 | Loss of function of ICa. |
Like most inherited channelopathies, SQTC is genetically heterogeneous with an autosomal dominant mode of inheritance. Both gain-of-function mutations in potassium channels and loss-of-function mutations in calcium channels have been implicated in the genetic basis of the disease.81To the present day, pathogenic variants have been identified in five different genes that are responsible for subtypes of the syndrome ranging from SQTC1 to SQTC52.
The diagnosis of SQTS should be considered in patients who mainly have an EKG study that reveals a short QT interval, family history of SQTS, or sudden death before 40 years old.82
Brugada syndrome (BrS)
Brugada syndrome (BrS), also known as right bundle branch block syndrome, persistent ST-segment elevation, and sudden death syndrome, is a hereditary channelopathy first described in 1992, which is mainly characterized by a characteristic electrocardiographic pattern in the right precordial leads and the predisposition to present ventricular arrhythmias and sudden cardiac death.83It is considered the cause of 4% - 12% of all sudden deaths and up to 20% of sudden deaths that occur in patients with structurally normal hearts.84
It has been determined that BrS has a prevalence of 5 in every 10,000 inhabitants, although this figure possibly underestimates the real prevalence given that patients may present silent forms of the disease.85In certain populations of Southeast Asia, such as the Philippines, Japan and Thailand are considered an endemic health problem.85
Although BrS is transmitted under an autosomal dominant pattern of inheritance84, the estimated clinical penetrance based on the analysis of individuals carrying pathogenic variants of the sodium channel is 16%.84Despite the fact that to date several genes have been identified that cause BrS, in most cases (~ 65%) no genetic mutation is identified.2
In 1998, the first pathogenic variants related to BrS were identified in the SCN5A gene (locus 3p21), which codes for the cardiac sodium channel.86The selection of said gene in a small number of families and individuals with idiopathic ventricular fibrillation and an EKG characteristic of BrS revealed missense, frameshift, and splicing variants. Pathogenic loss-of-function variants in SCN5A were found in up to 28% of patients with positive genetic tests.86
Currently, more than 100 different pathogenic variants responsible for BrS have been described in the SCN5A gene, the effect of which in all cases is the reduction of sodium transmembrane currents (INa), due to a quantitative reduction or a qualitative dysfunction of the channels. 85,86 In this subtype of BrS (Br1), the clinical findings that correlate with a pathogenic variant are a longer PR interval on resting ECG and an exaggerated increase in QRS duration with ventricular channel blocking agents. sodium.87Genetic background seems to play a very important role in the phenotypic expression of the disease in the ECG.88
Pathogenic variants in other genes have also been associated, less frequently, with the occurrence of BrS. Variants in the GPD1L (Br2) gene were initially identified by linkage analysis in a large family89and later in victims of sudden infant death syndrome.90On the other hand, in a group of patients with BrS associated with short QT intervals, candidate gene screening revealed loss-of-function mutations in CACNA1C resulting in Brugada syndrome type 3 (BrS3) and in CACNB2 resulting in Brugada syndrome type 4 (BrS4).90Other genes related to BrS less frequent (<1% of cases) are: SCN1B, KCNE3, SCN3B, HCN4.91
In patients with a positive Brugada pattern on ECG, genetic screening targeting the SCN5A gene may be helpful. In the event that a pathogenic variant is found, it is recommended to carry out specific mutation tests in first-degree relatives82. The finding of a genetic variant is an indicator of the potential development of the clinical phenotype of the disease. These patients should be carefully monitored in order to identify the spontaneous appearance of the Brugada type 1 pattern, the appearance of clinical symptoms, or a combination of both.2
Catecholaminergic polymorphic ventricular tachycardia (CPVT)
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a highly lethal form of the hereditary arrhythmogenic disease characterized mainly by the occurrence of polymorphic ventricular arrhythmias in the presence of catecholamine production (through physical exercise and/or stress) and in the absence of structural heart disease.92
CPVT is an infrequent entity (1: 10,000 inhabitants), but very important, due to the high risk of sudden death that it entails.92This figure could be underreported because the fact of not identifying any genetic mutation could lead to diagnoses such as idiopathic ventricular fibrillation.93In the absence of adequate treatment, it is estimated that up to 50% of affected patients will die suddenly before the age of 20. The average age of onset of the disease ranges from 6 and 10 years, although sporadic cases around 30 years have also been reported.93
CPVT should be suspected in all young patients, especially children or adolescents, who present syncope related to physical exercise or emotional stress, who do not have structural heart disease and whose electrocardiogram shows a normal QT interval.93In the year 2001 Priori and Lahat discovered the genetic cause of the disease by identifying two variants95, one inherited under an autosomal dominant pattern associated with variants in the gene that codes for the cardiac ryanodine receptor (RyR2) and another inherited under an autosomal dominant pattern. autosomal recessive associated with homozygous mutations in the gene encoding the cardiac isoform of calsequestrin (CASQ2). Today it is possible to confirm the diagnosis by performing different genetic tests on the genes known to be associated with the disease.96,97
Subsequently, mutations in the calsequestrin 2 (CASQ2) gene were identified in a patient with an autosomal recessive inheritance pattern of CPVT (CPVT2).95This was the second gene related to CPVT. Calsequestrin 2 is a crucial protein in the regulation of intracellular Ca2+. It constitutes the largest reservoir of this ion in the sarcoplasmic reticulum and also modulates its release through RyR2. When the capacity of calsequestrin 2 to store Ca2+ decreases, the concentrations of this free ion in the intercellular space increase, a fact that will determine the premature activation of RyR2 that will release more Ca2+ ions during systole.96
Approximately 30% - 40% of patients with CPVT have a family history of sudden death; a fact that supports the genetic origin of this disease.93To date, 130 mutations in RyR2 have been characterized, representing between 50-60% of all those identified in CPVT.96These exhibit a pattern of autosomal dominant inheritance, a penetrance of 83%, and condition type 1 VTCP.93Cases of patients with VTCP due to pathogenic variants in the CASQ2 gene that condition type 2 CPVT, represent only 5% of all cases. cases. They are inherited under an autosomal recessive pattern98, due to homozygous or compound heterozygous pathogenic variants in the CASQ2 gene and have a penetrance of 78%.99
Alterations in other genes have also been identified through genome-wide linkage analysis including the calmodulin 1 (CALM1) genes in the origin of CPVT type 4 and the triadin gene (TRDN) as a cause. of type 5 CPVT.100Finally, SCN5A has been implicated in exercise-induced polymorphic ventricular arrhythmias, with the recent identification of a new missense mutation in a highly conserved region of the gene in a large family with a CPVT phenotype.
CPVT should be suspected in all young patients (mainly children/adolescents) presenting with syncope related to physical exercise, emotional stress, in the absence of structural heart disease, and with an EKG without abnormalities (normal QT interval).93
The differential diagnosis should be made with other hereditary heart diseases that are susceptible to producing malignant ventricular arrhythmias due to exercise and/or emotional stress, such as long QT syndrome (LQTS 1, LQTS 2 and LQTS 3), since both conditions share clinical characteristics such as age debut, absence of structural heart disease, family history of syncope or sudden death, appearance of symptoms related to physical or emotional stress and the polymorphic nature of ventricular arrhythmias.93The biggest difference between both is the prolonged QT interval in LQTS, although a normal QT interval does not exclude the diagnosis of LQTS (5-10% of carriers of genetic variants in LQTS show a normal QT interval).
Genetic testing is recommended as part of the evaluation of an individual with exercise-induced or polymorphic ventricular tachycardia (documented on ECG), and when these arrhythmias occur in the setting of emotional stress.(100They may also be considered in the evaluation of cardiac arrest in the context of exertion or emotional stress. RyR2 gene sequencing is recommended in sporadic cases or when family history suggests autosomal dominant inheritance.(100In sporadic cases where there is a family history of consanguinity or when an autosomal recessive inheritance pattern is suspected, sequencing is recommended. CASQ2 gene sequencing. Pathogenic variants in the CASQ2 gene have been identified in only 1%-2% of all CPVT patients.100
CONCLUSIONS
The role of genetic studies in the risk stratification of sudden death is based on the study of the patient with clinical suspicion of a cardiac disorder with genetic origin. Genetic tests are useful for the diagnosis and clinical management of the patient, in addition, timely advice allows the identification of families at risk to establish personalized follow-up and prevention measures, based on clinical guidelines and relevant scientific information.
Although technological advances currently allow us to have genetic studies available through multigene panels through next-generation sequencing (NGS), and in turn allow the identification of new variants associated with the disease, not all Patients have hospital access to these tests. One limitation of this review was not having a national database on molecular studies of patients with heart disease of genetic origin, this could be due to the fact that they are not performed in any laboratory in the country, the cost of performing them abroad could not be covered by patients or that there have been no publications on this topic.
REFERENCES
1. Girolami F, Frisso G, Benelli M, Crotti L, Iascone M, Mango R, et al. Contemporary genetic testing in inherited cardiac disease: tools, ethical issues, and clinical applications. Journal of cardiovascular medicine (Hagerstown, Md). 2018;19(1):1. [ Links ]
2. Spears DA, Gollob MH. Genetics of inherited primary arrhythmia disorders. The application of clinical genetics. 2015;8:215. [ Links ]
3. Myerburg RJ. Sudden cardiac death: exploring the limits of our knowledge. Journal of cardiovascular electrophysiology. 2001;12(3):369-81. [ Links ]
4. Deo R, Albert CM. Epidemiology and genetics of sudden cardiac death. Circulation. 2012;125(4):620-37. [ Links ]
5. Teekakirikul P, Kelly MA, Rehm HL, Lakdawala NK, Funke BH. Inherited cardiomyopathies: molecular genetics and clinical genetic testing in the postgenomicera. J Mol Diagn. 2013 Mar;15(2):158-70. [ Links ]
6. Giudicessi JR, Kullo IJ, Ackerman MJ, editors. Precision cardiovascular medicine: state of genetic testing. Mayo Clinic Proceedings; 2017: Elsevier. [ Links ]
7. Giudicessi JR, Ackerman MJ. Genetic testing in heritable cardiac arrhythmia syndromes: differentiating pathogenic mutations from background genetic noise. Current opinion in cardiology. 2013;28(1):63. [ Links ]
8. Kauferstein S, Kiehne N, Jenewein T, Biel S, Kopp M, König R, et al. Genetic analysis of sudden unexplained death: a multidisciplinary approach. Forensic science international. 2013;229(1-3):122-7. [ Links ]
9. Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild D. Prevalence of hypertrophic cardiomyopathy in a general population of young adults: echocardiographic analysis of 4111 subjects in the CARDIA Study. Circulation 1995, 92:785-789 [ Links ]
10. Maron BJ, Casey SA, Hauser RG, Aeppli DM: Clinical course of hypertrophic cardiomyopathy with survival to advanced age. J Am Coll Cardiol 2003, 42:882-888 [ Links ]
11. Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, et al. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: Executive Summary: A Report of the American College of Cardiology Foundation/American Heart Association Task Force of Practice Guidelines. Circulation. 2011;124:2761-96. [ Links ]
12. Nagueh SF, Bachinski LL, Meyer D, Hill R, Zoghbi WA, Tam JW, et al. Tissue Doppler imaging consistently detects myocardial abnormalities in patients with hypertrophic cardiomyopathy and provides a novel means for an early diagnosis before and independently of hypertrophy. Circulation. 2001;104:128-30. [ Links ]
13. Niimura H, Patton KK, McKenna WJ, Soults J, Maron BJ, Seidman JG, Seidman CE. Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly. Circulation. 2002;105:446-51. [ Links ]
14. Seidman CE, Seidman JG: Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: a personal history. Circ Res 2011, 108:743-750 [ Links ]
15. Fatkin D, Graham RM: Molecular mechanisms of inherited cardiomyopathies. Physiol Rev 2002, 82:945-980 [ Links ]
16. Teekakirikul P, Eminaga S, Toka O, Alcalai R, Wang L, Wakimoto H, Nayor M, et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-b. J Clin Invest 2010, 120:3520-3529. [ Links ]
17. Callis TE, Jensen BC, Weck KE, Willis MS: Evolving molecular diagnostics for familial cardiomyopathies: at the heart of it all. Expert Rev Mol Diagn 2010, 10:329-351. [ Links ]
18. Ho CY: Genetics and clinical destiny: improving care in hypertrophic cardiomyopathy. Circulation 2010, 122:2430-2440. [ Links ]
19. Alfares AA, Kelly MA, McDermott G, Funke BH, Lebo MS, Baxter SB, et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med. 2015;17:880-8. [ Links ]
20. Vandenberg JI, Perry MD, Perrin MJ, Mann SA, Ke Y, Hill AP. hERG K+ channels: structure, function, and clinical significance. Physiological reviews. 2012;92(3):1393-478. [ Links ]
21. MacCormick JM, McAlister H, Crawford J, French JK, Crozier I, Shelling AN, et al. Misdiagnosis of long QT syndrome as epilepsy at first presentation. Annals of emergency medicine. 2009;54(1):26-32. [ Links ]
22. Viskin S. Long QT syndromes and torsade de pointes. The Lancet. 1999;354(9190):1625-33. [ Links ]
23. Hershberger RE, Kushner JD, and Parks SB: Dilated Cardiomyopathy Overview. In GeneReviews [Internet]. Copyright University of Washington, Seattle. 1993e2012. Available at http://www.ncbi.nlm. nih.gov/books/NBK1309, last revised March 19, 2009 [ Links ]
24. Dec GW, Fuster V: Idiopathic dilated cardiomyopathy. N Engl J Med 1994, 331:1564-1575. [ Links ]
25. Morita H, Seidman J, Seidman CE: Genetic causes of human heart failure. J Clin Invest 2005, 115:518-526. [ Links ]
26. Watkins H, Ashrafian H, Redwood C: Inherited cardiomyopathies. N Engl J Med 2011, 364:1643-1656. [ Links ]
27. Hershberger RE, Norton N, Morales A, Li D, Siegfried JD, Gonzalez- Quintana J: Coding sequence rare variants identified inMYBPC3,MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ Cardiovasc Genet 2010, 3:155-161. [ Links ]
28. Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S, Peterson A, Li D, Jakobs P, Litt M, Porter CB, Rahko PS, Hershberger RE: Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am Heart J 2008, 156:161-169. [ Links ]
29. Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, et al.Truncations of titin causing dilated cardiomyopathy. N Engl J Med 2012, 366:619-628. [ Links ]
30. Hershberger RE, Lindenfeld J, Mestroni L, Seidman CE, Taylor MR, Towbin JA. Genetic evaluation of cardiomyopathy--a Heart Failure Society of America practice guideline. J Card Fail. 2009b;15:83-97. [ Links ]
31. Elkayam U, Akhter MW, Singh H, Khan S, Bitar F, Hameed A, Shotan A. Pregnancy-associated cardiomyopathy: clinical characteristics and a comparison between early and late presentation. Circulation. 2005;111:2050-5. [ Links ]
32. Hershberger RE, Siegfried JD. State of the Art Review. Update 2011: clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2011;57:1641-9. [ Links ]
33. Hershberger RE, Parks SB, Kushner JD, Li D, Ludwigsen S, Jakobs P, Nauman D, Burgess D, Partain J, Litt M. Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy. Clin Transl Sci. 2008;1:21-6. [ Links ]
34. Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, Baxter SM, Bowser M, Harrison B, Aaron D, Mahanta LM, Lakdawala NK, McDermott G, White ET, Rehm HL, Lebo M, Funke BH. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med. 2014;16:601-8. [ Links ]
35. Caleshu C, Day S, Rehm HL, Baxter S. Use and interpretation of genetic tests in cardiovascular genetics. Heart. 2010;96:1669-75. [ Links ]
36. Morales A, Hershberger RE. The rationale and timing of molecular genetic testing for dilated cardiomyopathy. Can J Cardiol. 2015;31:1309-12. [ Links ]
37. Corrado D, Basso C, Thiene G, McKenna WJ, Davies MJ, Fontaliran F, Nava A, Silvestri F, Blomstrom-Lundqvist C, Wlodarska EK, Fontaine G, Camerini F: Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J Am Coll Cardiol 1997, 30:1512-1520. [ Links ]
38. Bauce B, Frigo G, Marcus FI, Basso C, Rampazzo A, Maddalena F, Corrado D, Winnicki M, Daliento L, Rigato I, Steriotis A, Mazzotti E, Thiene G, Nava A: Comparison of clinical features of arrhythmogenic right ventricular cardiomyopathy in men versus women. Am J Cardiol 2008, 102:1252-1257. [ Links ]
39. Lombardi R, Marian AJ: Molecular genetics and pathogenesis of arrhythmogenic right ventricular cardiomyopathy: a disease of cardiac stem cells. Pediatr Cardiol 2011, 32:360-365. [ Links ]
40. den Haan AD, Tan BY, Zikusoka MN, Llado LI, Jain R, Daly A, Tichnell C, James C, Amat-Alarcon N, Abraham T, Russell SD, Bluemke DA, Calkins H, Dalal D, Judge DP: Comprehensive desmosome mutation analysis in North Americans with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Cardiovasc Genet 2009, 2:428-435. [ Links ]
41. Protonotarios N, Tsatsopoulou A: Naxos disease and Carvajal syndrome: cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Pathol 2004, 13:185-194. [ Links ]
42. Merner ND, Hodgkinson KA, Haywood AF, Connors S, French VM, Drenckhahn JD, Kupprion C, Ramadanova K, Thierfelder L, McKenna W, Gallagher B, Morris-Larkin L, Bassett AS, Parfrey PS, Young TL: Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet 2008, 82:809-821. [ Links ]
43. Oechslin E, Jenni R: Left ventricular non-compaction revisited: a distinct phenotype with genetic heterogeneity? Eur Heart J 2011, 32: 1446-1456. [ Links ]
44. Pignatelli RH, McMahon CJ, Dreyer WJ, Denfield SW, Price J, Belmont JW, Craigen WJ, Wu J, El Said H, Bezold LI, Clunie S, Fernbach S, Bowles NE, Towbin JA: Clinical characterization of left ventricular noncompaction in children: a relatively common form of cardiomyopathy. Circulation 2003, 108:2672-2678. [ Links ]
45. Ritter M, Oechslin E, Sutsch G, Attenhofer C, Schneider J, Jenni R: Isolated noncompaction of the myocardium in adults. Mayo Clin Proc 1997, 72:26-31. [ Links ]
46. Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O'Connell J, Olsen E, Thiene G, Goodwin J, Gyarfas I, Martin I, Nordet P: Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 1996, 93:841-842. [ Links ]
47. Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kuhl U, Maisch B, McKenna WJ, Monserrat L, Pankuweit S, Rapezzi C, Seferovic P, Tavazzi L, Keren A: Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2008, 29:270-276. [ Links ]
48. Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB: Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006, 113:1807-1816. [ Links ]
49. Finsterer J: Cardiogenetics, neurogenetics, and pathogenetics of left ventricular hypertrabeculation/noncompaction. Pediatr Cardiol 2009, 30:659-681. [ Links ]
50. Vatta M, Mohapatra B, Jimenez S, Sanchez X, Faulkner G, Perles Z, et al. Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. J Am Coll Cardiol 2003, 42:2014-2027. [ Links ]
51. Hermida-Prieto M, Monserrat L, Castro-Beiras A, Laredo R, Soler R, Peteiro J, Rodriguez E, Bouzas B, Alvarez N, Muniz J, Crespo- Leiro M: Familial dilated cardiomyopathy and isolated left ventricular noncompaction associated with lamin A/C gene mutations. Am J Cardiol 2004, 94:50-54. [ Links ]
52. Ichida F, Tsubata S, Bowles KR, Haneda N, Uese K, Miyawaki T, Dreyer WJ, Messina J, Li H, Bowles NE, Towbin JA: Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome. Circulation 2001, 103:1256-1263. [ Links ]
53. Ichida F. Left ventricular noncompaction. Circ J 2009, 73:19-26. [ Links ]
54. Sen-Chowdhry S, Syrris P, McKenna WJ: Genetics of restrictive cardiomyopathy. Heart Fail Clin 2010, 6:179-186. [ Links ]
55. Karam S, Raboisson MJ, Ducreux C, Chalabreysse L, Millat G, Bozio A, Bouvagnet P: A de novo mutation of the beta cardiac myosin heavy chain gene in an infantile restrictive cardiomyopathy. Congenit Heart Dis 2008, 3:138-143. [ Links ]
56. Kubo T, Gimeno JR, Bahl A, Steffensen U, Steffensen M, Osman E, Thaman R, Mogensen J, Elliott PM, Doi Y, McKenna WJ: Prevalence, clinical significance, and genetic basis of hypertrophic cardiomyopathy with restrictive phenotype. J Am Coll Cardiol 2007, 49:2419-2426. [ Links ]
57. Arbustini E, Pasotti M, Pilotto A, Pellegrini C, Grasso M, Previtali S, Repetto A, Bellini O, Azan G, Scaffino M, Campana C, Piccolo G, Vigano M, Tavazzi L: Desmin accumulation restrictive cardiomyopathy and atrioventricular block associated with desmin gene defects. Eur J Heart Fail 2006, 8:477-483. [ Links ]
58. Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart rhythm. 2011;8(8):1308-39. [ Links ]
59. Priori S, Cantu F, Schwartz P. The long QT syndrome: new diagnostic and therapeutic approach in the era of molecular biology. Schweizerische medizinische Wochenschrift. 1996;126(41):1727-31. [ Links ]
60. Viskin S, Rosovski U, Sands AJ, Chen E, Kistler PM, Kalman JM, et al. Inaccurate electrocardiographic interpretation of long QT: the majority of physicians cannot recognize a long QT when they see one. Heart Rhythm. 2005;2(6):569-74. [ Links ]
61. Towbin JA, Vatta M. Molecular biology and the prolonged QT syndromes. The American journal of medicine. 2001;110(5):385-98. [ Links ]
62. Tristani-Firouzi M, Chen J, Mitcheson JS, Sanguinetti MC. Molecular biology of K+ channels and their role in cardiac arrhythmias. The American journal of medicine. 2001;110(1):50-9. [ Links ]
63. Muñoz Castellano J. Síndrome de QT largo y Torsade de Pointes. Emergencias. 2004;16:85-92. [ Links ]
64. Khan IA. Clinical and therapeutic aspects of congenital and acquired long QT syndrome. The American journal of medicine. 2002;112(1):58-66. [ Links ]
65. Berge K, Haugaa K, Früh A, Anfinsen OG, Gjesdal K, Siem G, et al. Molecular genetic analysis of long QT syndrome in Norway indicating a high prevalence of heterozygous mutation carriers. Scandinavian journal of clinical and laboratory investigation. 2008;68(5):362-8. [ Links ]
66. Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, et al. Spectrum of mutations in long-QT syndrome genes: KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102(10):1178-85. [ Links ]
67. Goldenberg I, Moss AJ, Bradley J, Polonsky S, Peterson DR, McNitt S, et al. Long-QT syndrome after age 40. Circulation. 2008;117(17):2192-201. [ Links ]
68. Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, et al. Risk stratification in the long-QT syndrome. New England Journal of Medicine. 2003;348(19):1866-74. [ Links ]
69. Spazzolini C, Mullally J, Moss AJ, Schwartz PJ, McNitt S, Ouellet G, et al. Clinical implications for patients with long QT syndrome who experience a cardiac event during infancy. Journal of the American College of Cardiology. 2009;54(9):832-7. [ Links ]
70. Pagon R, Adam M, Ardinger H, Wallace S, Amemiya A, Bean L, et al. GeneReviews(r)[Internet]. Seattle (WA): University of Washington, Seattle. 1993;201. [ Links ]
71. Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Robinson JL, Priori SG, Benhorin J, Locati EH, Towbin JA, Keating MT, Lehmann MH, Hall WJ. Influence of genotype on the clinical course of the long-QT syndrome. International Long-QT Syndrome Registry Research Group. N Engl J Med. 1998;339:960-5. [ Links ]
72. Goldenberg I, Zareba W, Moss AJ. Long QT syndrome. Curr Probl Cardiol. 2008;33:629-94. [ Links ]
73. Nannenberg EA, Sijbrands EJ, Dijksman LM, Alders M, van Tintelen JP, Birnie M, Van Langen IM, Wilde AA. Mortality of inherited arrhythmia syndromes: insight into their natural history. Circ Cardiovasc Genet. 2012;5:183-9. [ Links ]
74. Schwartz PJ, Priori SG, Napolitano C. How really rare are rare diseases? J Cardiovasc Electrophysiol. 2003;14:1120-1 [ Links ]
75. Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, Vicentini A, Spazzolini C, Nastoli J, Bottelli G, Folli R, Cappelletti D. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348:1866-74. [ Links ]
76. Goldenberg I, Horr S, Moss AJ, Lopes CM, Barsheshet A, McNitt S, Zareba W, Andrews ML, Robinson JL, Locati EH, Ackerman MJ, Benhorin J, Kaufman ES, Napolitano C, Platonov PG, Priori SG, Qi M, Schwartz PJ, Shimizu W, Towbin JA, Vincent GM, Wilde AA, Zhang L. Risk for life-threatening cardiac events in patients with genotype-confirmed long-QT syndrome and normal-range corrected QT intervals. J Am Coll Cardiol. 2011;57:51-9. [ Links ]
77. Khera S, Jacobson JT. Short QT syndrome in current clinical practice. Cardiology in review. 2016;24(4):190-3. [ Links ]
78. Gussak I, Brugada P, Brugada J, Wright RS, Kopecky SL, Chaitman BR, et al. Idiopathic short QT interval: a new clinical syndrome? Cardiology. 2000;94(2):99-102. [ Links ]
79. Campuzano Ó, Sarquella-Brugada G, Brugada R, Brugada P, Brugada J. Bases genéticas de las arritmias malignas y las miocardiopatías. Revista española de cardiología. 2009;62(4):422-36. [ Links ]
80. Patel C, Yan G-X, Antzelevitch C. Short QT syndrome: from bench to bedside. Circulation: Arrhythmia and Electrophysiology. 2010;3(4):401-8. [ Links ]
81. Giustetto C, Schimpf R, Mazzanti A, Scrocco C, Maury P, Anttonen O, et al. Long-term follow-up of patients with short QT syndrome. Journal of the American College of Cardiology. 2011;58(6):587-95. [ Links ]
82. Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart rhythm. 2013;10(12):1932-63. [ Links ]
83. Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. Journal of the American College of Cardiology. 1992;20(6):1391-6. [ Links ]
84. Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111(5):659-70. [ Links ]
85. Hermida JS, Lemoine JL, Aoun FB, Jarry G, Rey JL, Quiret JC. Prevalence of the brugada syndrome in an apparently healthy population. The American journal of cardiology. 2000;86(1):91-4. [ Links ]
86. Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392(6673):293-6. [ Links ]
87. Smits JP, Eckardt L, Probst V, Bezzina CR, Schott JJ, Remme CA, et al. Genotype-phenotype relationship in Brugada syndrome: electrocardiographic features differentiate SCN5A-related patients from non-SCN5A-related patients. Journal of the American College of Cardiology. 2002;40(2):350-6. [ Links ]
88. Probst V, Wilde AA, Barc J, Sacher F, Babuty D, Mabo P, et al. SCN5A mutations and the role of genetic background in the pathophysiology of Brugada syndrome. Circulation Cardiovascular genetics. 2009;2(6):552-7 [ Links ]
89. Weiss R, Barmada MM, Nguyen T, Seibel JS, Cavlovich D, Kornblit CA, et al. Clinical and molecular heterogeneity in the Brugada syndrome: a novel gene locus on chromosome 3. Circulation. 2002;105(6):707-13. [ Links ]
90. Van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, et al. Molecular and functional characterization of novel glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) mutations in sudden infant death syndrome. Circulation. 2007;116(20):2253-9. [ Links ]
91. Burashnikov E, Pfeiffer R, Barajas-Martinez H, Delpon E, Hu D, Desai M, et al. Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart rhythm. 2010;7(12):1872-82. [ Links ]
92. Marks AR, Priori S, Memmi M, Kontula K, Laitinen PJ. Involvement of the cardiac ryanodine receptor/calcium release channel in catecholaminergic polymorphic ventricular tachycardia. Journal of cellular physiology. 2002;190(1):1-6. [ Links ]
93. Cruz Cardentey M. Taquicardia ventricular polimórfica catecolaminérgica. Revista Cubana de Investigaciones Biomédicas. 2012;31(2):187-98. [ Links ]
94. Cohen TJ, Liem LB, Hancock EW. Association of bidirectional ventricular tachycardia with familial sudden death syndrome. The American journal of cardiology. 1989;64(16):1078-9. [ Links ]
95. Lahat H, Pras E, Eldar M. A missense mutation in CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Annals of medicine. 2004;36 Suppl 1:87-91. [ Links ]
96. Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103(2):196-200. [ Links ]
97. Eldar M, Pras E, Lahat H. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Cold Spring Harbor symposia on quantitative biology. 2002;67:333-7. [ Links ]
98. Lahat H, Pras E, Olender T, Avidan N, Ben-Asher E, Man O, et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. American journal of human genetics. 2001;69(6):1378-84. [ Links ]
99. Postma AV, Denjoy I, Kamblock J, Alders M, Lupoglazoff JM, Vaksmann G, et al. Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients. Journal of medical genetics. 2005;42(11):863-70. [ Links ]
100. Roux-Buisson N, Cacheux M, Fourest-Lieuvin A, Fauconnier J, Brocard J, Denjoy I, et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Human molecular genetics. 2012;21(12):2759-67. [ Links ]
8 Article published by the Journal of the faculty of Human Medicine of the Ricardo Palma University. It is an open access article, distributed under the terms of the Creatvie Commons license: Creative Commons Attribution 4.0 International, CC BY 4.0(https://creativecommons.org/licenses/by/1.0/), that allows non-commercial use, distribution and reproduction in any medium, provided that the original work is duly cited. For commercial use, please contact revista.medicina@urp.edu.pe.
Received: May 14, 2022; Accepted: August 17, 2022
 Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
