











 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Se considera que una muerte repentina e inexplicable se debe a un trastorno arrítmico primario cuando no se encuentra una enfermedad cardíaca estructural en la necropsia y no hay documentación previa de enfermedad cardíaca.1,2En la población general, la incidencia de muerte súbita varía de 1/100,000 en adolescentes a 1/1,000 en individuos de 45 a 75 años.3Se cree que las canalopatías son responsables del 10% al 15% de los casos de muerte súbita sin explicación en adultos jóvenes y niños.4
Las cardiomiopatías hereditarias son un grupo genético y fenotípicamente heterogéneo que predispone a insuficiencia cardíaca y muerte súbita, debido a variantes patogénicas en genes que codifican proteínas fundamentales en los componentes estructurales de cardiomiocitos y son clasificadas funcional y morfológicamente por sus características en: miocardiopatía hipertrófica, miocardiopatía dilatada, miocardiopatía/displasia arritmogénica ventricular derecha, miocardiopatía por ventrículo no compactado y cardiomiopatía restrictiva.5
Las canalopatías son un conjunto de síndromes hereditarios de arritmia cardíaca, de expresividad clínica variable, que incluye al síndrome de Brugada, la taquicardia ventricular polimórfica catecolaminérgica, el síndrome de QT largo, el síndrome de QT corto y el síndrome de repolarización precoz.6Estas canalopatías surgen de defectos en complejos macromoleculares de canales iónicos o proteínas críticas para el manejo intracelular del calcio, sodio y/o potasio. Los pacientes que poseen una canalopatía suelen tener un corazón estructuralmente normal, pero estar predispuestos a presentar síncope/crisis arrítmicas y muerte súbita cardíaca.7
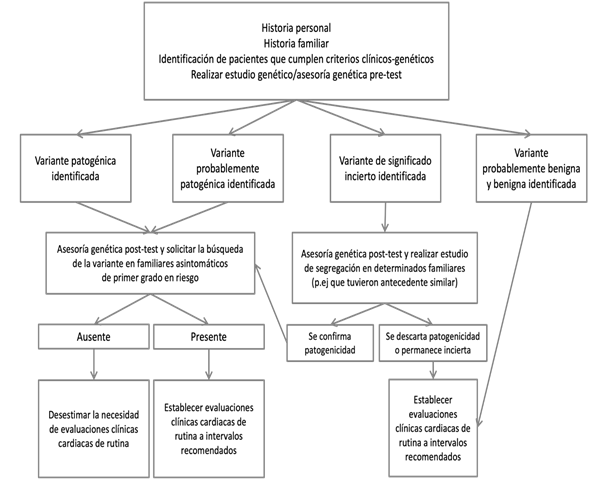
Desde el descubrimiento de los genes asociados a la enfermedad cardiovascular hereditaria de origen monogénico a principios de los años 90 hasta la actualidad donde se realizan estudios de paneles genéticos o exomas completos6en pacientes con sospecha de una cardiopatía hereditaria, los estudios genéticos cumplen un rol importante en apoyo al diagnóstico de un trastorno de arritmia primaria. Además, proporcionan información pronóstica y permiten ampliar la evaluación a familiares de primer grado asintomáticos en riesgo. Los estudios genéticos son utilizados en la práctica clínica al momento de la estratificación de riesgos y manejo de los pacientes, basado en una medicina de precisión, enfocada en la medicina genómica (Figura 1)8
El objetivo de esta revisión es poner en evidencia la importancia del asesoramiento genético de los pacientes con enfermedad cardiovascular hereditaria y su evaluación a través de un equipo multidisciplinario.
Cardiomiopatía hipertrófica
La cardiomiopatía hipertrófica (CMH) se caracteriza por la hipertrofia ventricular izquierda en ausencia de una afección sistémica subyacente u otra enfermedad cardíaca, como enfermedad cardíaca valvular o hipertensión. Se estima que posee una prevalencia de 1 en 500 en la población general.9La CMH es la afección cardíaca de tipo hereditaria más común y la causa principal de muerte súbita en adultos jóvenes y atletas competitivos en Estados Unidos.10Aunque la edad de inicio de la CMH puede variar desde la infancia hasta la vejez, las manifestaciones, en aquellos portadores de una variante patogénica, generalmente no aparecen antes de la adolescencia.10La CMH se hereda principalmente bajo un patrón autosómico dominante, con penetrancia reducida y variabilidad clínica. Las manifestaciones clínicas varían desde ser completamente asintomáticos hasta insuficiencia cardíaca progresiva y muerte súbita causadas por defectos mecánicos o eléctricos.
La CMH se define por la presencia de hipertrofia ventricular izquierda sin explicación, con un espesor máximo de pared ≥15 mm en adultos o un score Z > 3 en niños.11Si existen antecedentes familiares de CMH, o si las pruebas genéticas confirman que un familiar ha heredado alguna variante patogénica, el grosor máximo de la pared del VI ≥ 13 mm respaldará el diagnóstico. La alteración en la relajación del ventrículo izquierdo, puede identificarse en individuos que posean una variante patogénica en un gen que codifica un componente del sarcómero (CMH no sindrómica) que tienen un grosor normal de la pared del ventrículo izquierdo12lo que sugiere que la disfunción diastólica es un fenotipo temprano de CMH en lugar de una consecuencia secundaria de la hipertrofia del ventrículo izquierdo. Aunque la hipertrofia del ventrículo izquierdo y un diagnóstico clínico de CMH con frecuencia son evidentes durante la adolescencia, alrededor del inicio de la pubertad o durante la edad adulta, el inicio puede ser más temprano (en la infancia y la niñez).13
La CMH se describe con frecuencia como una enfermedad del sarcómero, y se han detectado variantes patogénicas en casi todas las proteínas sarcoméricas.(14Los mecanismos moleculares incluyen una mayor actividad de ATPasa activada por actina, interrupción de la interacción actina-miosina y una alteración en la señalización intracelular del calcio en los cardiomiocitos.15Además, algunos datos sugieren que la hipertrofia del ventrículo izquierdo puede ser causada por alteraciones en las vías de señalización del factor de crecimiento transformante B y CaMKII Mef2.16
De acuerdo a la etiología genética de la CMH, se han descrito variantes patogénicas en diversos genes que codifican las proteínas sarcoméricas, siendo la mayoría (80%) en los genes MYH7 y MYBPC3.17(Tabla 1) Las variantes patogénicas en los genes que codifican un componente del sarcómero se encuentran en aproximadamente el 50% - 60% de los probandos (adultos y niños) con antecedentes familiares de CMH, y aproximadamente en el 20% -30% de los probandos sin antecedentes familiares de HCM.18.
Aproximadamente el 3% - 5% de las personas afectadas tienen variantes bialélicas en 1 gen o variantes heterocigotas en más de 1 gen.19Recientemente, el espectro de genes asociados a la CMH se ha ampliado a genes no sarcoméricos e incluye genes que codifican proteínas del disco Z y proteínas ubicadas en el retículo sarcoplásmico y membrana plasmática. Sin embargo, las variantes en estos genes son poco frecuentes.
Las variantes en el gen MYH7 generalmente conducen a una hipertrofia ventricular izquierda significativa, en la segunda década de la vida y se cree que están asociadas con un mayor riesgo de insuficiencia cardíaca y muerte súbita cardiaca.20En contraste, se cree que las variantes patogénicas en MYBPC3 se asocian con una edad mayor al diagnóstico18aunque también se han identificado en una proporción significativa de pacientes con hipertrofia del ventrículo izquierdo de inicio en la infancia.19
El diagnóstico diferencial de la CMH incluye diversos síndromes que generalmente se manifiestan con compromiso multiorgánico pero que también pueden presentarse con hipertrofia del ventrículo izquierdo de forma aislada o predominante. Estos síndromes incluyen cardiomiopatías metabólicas de depósito como la enfermedad de Danon (gen LAMP2), el síndrome de Wolff-Parkinson-White (gen PRKAG2)21, la enfermedad de Fabry que es un trastorno de almacenamiento lisosomal (gen GLA)22así como trastornos sindrómicos (con otra afectación sistémica) (Tabla 2) y no sindrómicos (sin otra afectación sistémica).
Tabla 1. Genes asociados a cardiomiopatía hipertrófica .
| Gen | Herencia | % de CMH causada por variante patogénica en este gen | Desordenes alélicos | OMIM (Online Mendelian Inherited in Man) |
|---|---|---|---|---|
| MYBPC3 | AD | 50% | CMD | 600958 |
| MYH7 | AD | 33% | Miopatía por depósito de miosina Cardiomiopatía por ventrículo no compactado | 160760 |
| TNNI3 | AD | 5% | CMD Cardiomiopatía restrictiva | 191044 |
| TNNT2 | AD | 4% | CMD Cardiomiopatía por ventrículo no compactado Cardiomiopatía restrictiva familiar | 191045 |
| ACTC1 | AD | <3% | CMD | 102540 |
| MYL2 | AD | <3% | 160781 | |
| MYL3 | AD AR | <3% | 160790 | |
| TPM1 | AD | <3% | CMD | 191010 |
| PLN | AD | <3% | CMD | 172405 |
| ALPK3 | AR | Raro | 617608 | |
| ACTN2 | AD | <1% | CMD | 102573 |
| CSRP3 | AD | <1% | CMD | 600824 |
| TNNC1 | AD | <1% | CMD | 191040 |
| JPH2 | AD | Raro | 605267 | |
| MYOZ2 | AD | <1% | 605602 | |
| NEXN | AD | <1% | CMD | 613121 |
| ANKRD1 | AD | Raro | ||
| CALR3 | AD | Raro | 611414 | |
| KLF10 | AD | Raro | 601878 | |
| MYH6 | AD | Raro | CMD | 160710 |
| MYLK2 | Digénica | Raro | 606566 | |
| MYOM1 | AD | Raro | 603508 | |
| MYPN | AD | Raro | CMD Miopatía nemalínica | 608517 |
| OBSCN | AD | Raro | 608616 | |
| PDLIM3 | AD | Raro | 605889 | |
| RYR2 | AD | Raro | Cardiomiopatía arritmogénica del ventrículo derecho Taquicardia ventricular polimórfica catecolaminérgica | 180902 |
| TCAP | AD | Raro | CMD | 604488 |
| TRIM63 | AD | Raro | 606131 | |
| TTN | AD | Raro | CMD Miopatía hereditaria con falla respiratoria temprana Miopatía distal de Salih Miopatía distal Udd | 613765 |
| VCL | AD | Raro | CMD | 193065 |
AD: Autosómico dominante; AR: Autosómico recesiva; CMD: Cardiomiopatía dilatada.
Tabla 2. Cardiomiopatía hipertrófica sindrómica.
| Desorden | Gen(es) | Herencia | Otras características clínicas (no cardiacas) |
|---|---|---|---|
| Enfermedad de Danon | LAMP2 | XL | · Miopatía · Distrofia retinal |
| Enfermedad de Fabry | GLA | XL | · Crisis periódicas de dolor en las extremidades · Angioqueratomas · Hipohidrosis · Anomalías oculares · Proteinuria y deterioro de función renal |
| Ataxia deFriedreich | FXN | AR | · Ataxia, lentamente progresiva antes de los 25 años · Disartria · Debilidad muscular |
| Enfermedad de almacenamiento de glucógeno del corazón, congénita letal | PRKAG2 | AD | · Hipoglicemia neonatal · Miopatía vacuolar · Dismorfia facial y/o macroglosia |
| Amiloidosis hereditaria por transtiretina | TTR | AD | · Neuropatía sensitivomotora periférica progresiva lenta y neuropatía autonómica · Opacidad vítrea · Amiloidosis del SNC |
| Enfermedad de Pompe | GAA | AR | · Mala alimentación · Macroglosia · Retraso motor / debilidad muscular · Dificultad respiratoria |
| Rasopatías: Síndrome Noonan Síndrome Cardiofasciocutáneo Síndrome Costello Síndrome de Noonan con multiples lentigines | BRAF HRAS KRAS LZTR1 MAP2K1 MAP2K2 NRAS PTPN11 RAF1 RASA2 RRAS RIT1 SOS1 SOS2 | AD | · Facies características · Talla baja · Retraso variable del desarrollo variable · Cuello ancho y alado |
AD: Autosómico dominante; AR: Autosómico recesiva; XL: Ligado al cromosoma X
Cardiomiopatía dilatada
La cardiomiopatía dilatada (CMD) se define por dilatación del ventrículo izquierdo y disfunción sistólica, y es la indicación más común para el trasplante cardíaco en los Estados Unidos.23El espectro de manifestaciones clínicas incluye insuficiencia cardíaca, tromboembolismo y muerte súbita. La prevalencia estimada de CMD de tipo idiopática es de 1 de cada 2500 individuos. El porcentaje de CMD idiopáticas que tienen una etiología genética se estima entre 30% y 50% en función de la presencia de antecedentes familiares.23La edad de inicio puede variar desde la infancia hasta la edad adulta tardía, aunque la mayoría de los pacientes son diagnosticados entre 20 y 50 años de edad.24En comparación con la CMH, que es principalmente una enfermedad del sarcómero, la CMD presenta una mayor heterogeneidad genética (más de 40 genes descritos hasta la fecha) .25
La CMD se hereda principalmente bajo un patrón autosómico dominante, sin embargo se han descrito también por herencia ligada al cromosoma X.26Debido a la heterogeneidad de locus y alélica que posee la CMD, las pruebas genéticas que se utilizan con mayor frecuencia son los paneles genéticos. Se estima que se pueden identificar variantes patogénicas en 17% a 30% de los individuos con CMD cuando se realizan análisis por paneles multigenes de hasta 20 genes.27Las variantes patogénicas en los genes LMNA y SCN5 están claramente asociadas a CMD y enfermedad del sistema de conducción.28Las variantes patogénicas en el gen TTN se identifican hasta en el 25% de los casos familiares y el 18% de casos esporádicos de CMD.29(Tabla 3).
Las personas con CMD en las que se ha determinado que la causa no es adquirida (secundaria) o sindrómica (con otra afectación sistémica) (Tabla 4), tienen CMD no sindrómica (sin otra afectación sistémica).
Tabla 3. Genes asociados a cardiomiopatía dilatada.
| Gen | Herencia | % de CMH causada por variante patogénica en este gen | Desordenes alélicos | OMIM (Online Mendelian Inherited in Man) | Gen |
|---|---|---|---|---|---|
| ACTC1 | <1% | AD | CMH familiar | 102540 | |
| ACTN2 | <1% | AD | CMH familiar | 102573 | |
| ANKRD1 | 2.2% | AD | 609599 | ||
| BAG3 | 2.5% | AD | Miopatía miofibrilar progresiva | 603883, PS601419 | |
| CSRP3 | <1% | AD | CMH familiar | 600824 | |
| DES | <1% | AD | Arritmia y afectación neuromuscular | Miopatía miofibrilar | 125660, PS601419 |
| DMD | ? | XL | Afectación neuromuscular | Distrofinopatías | 300377 |
| DSG2 | AD | Posible afectación del ventrículo derecho | 125671 | ||
| EYA4 | ? | AD | Sordera | Sordera no sindrómica | 603550 |
| LDB3 | 1% | AD | Miopatía miofibrilar | 605906, PS601419 | |
| LMNA | 6% | AD | Arritmia y enfermedad del sistema de conducción | Lipodistrofia parcial Charcot Marie Tooth 2B1 Distrofia muscular Emery-Dreifuss Síndrome Progeria de Hutchinson-Gilford Síndrome de Werner atípico Enfermedad muscular relacionada a LMNA | 150330 |
| MYBPC3 | 2%-4% | AD | CMH familiar | 600958 | |
| MYH6 | 3%-4% | AD | CMH familiar | 160710 | |
| MYH7 | 4.2% | AD | Miopatía distal de Laing CMH familiar Miopatía por depósito (MYH7) Cardiomiopatía por ventrículo no compactado | 160760 | |
| NEXN | <1% | AD | CMH familiar | 613121 | |
| PLN | ? | AD | Arritmia y enfermedad del sistema de conducción | 172405 | |
| PSEN1 | <1% | AD | Enfermedad de Alzheimer de inicio temprano | 104311 | |
| PSEN2 | <1% | AD | Enfermedad de Alzheimer de inicio temprano y tardío | 600759 | |
| RBM20 | 1.9% | AD | Arritmia y enfermedad del sistema de conducción | 613171 | |
| SCN5A | 2%-4% | AD | Arritmia y enfermedad del sistema de conducción | Síndrome QT largo tipo 3 Síndrome de Brugada Fibrilación ventricular idiopática Síndrome del seno enfermo Enfermedad del sistema de conducción cardiaca | 600163 |
| SGCD | <1% | AD | Delta sarcoglicanopatia | 601411 | |
| TAZ | ? | XL | Presentación en la infancia | Síndrome de Barth Fibroelastosis endocardial tipo 2 Cardiomiopatía por ventrículo no compactado familiar | 300394 |
| TCAP | 1% | AD | CMH familiar | 604488 | |
| TMPO | 1.1% | AD | 188380 | ||
| TNNC1 | <1%-1.3% | AD | CMH familiar | 191040 | |
| TNNI3 | 1.3% (AD) <1% (AR) | AD | CMH familiar Cardiomiopatía restrictiva | 191044 | |
| TNNT2 | 2.9% | AD | CMH familiar Cardiomiopatía por ventrículo no compactado Cardiomiopatía restrictiva familiar relacionado a TNNT2 | 191045 | |
| TPM1 | <1%-1.9% | AD | CMH familiar | 191010 | |
| TTN 4 | 10%-20% | AD | Miopatía proximal, con compromiso muscular respiratorio temprano. Distrofia muscular tibial | 188840 | |
| VCL | 1% | AD | 193065 |
Tabla 4. Cardiomiopatía dilatada sindrómica.
| Desorden | Gen(es) | Herencia | Otras características clínicas (no cardiacas) | Observaciones |
|---|---|---|---|---|
| Síndrome de Barth | TAZ | XL | Neutropenia Debilidad muscular Retraso del desarrollo | |
| Síndrome de Carvajal | DSP | AR | Cabello lanoso Queratodermia palmoplantar | |
| Distrofia muscular de Duchenne / Becker | DMD | XL | En varones: Debilidad muscular Niveles séricos de CK elevados Pérdida de la deambulación en la infancia | Las mujeres heterocigotas pueden presenter CMD aislada |
| Distrofia muscular de Emery-Dreifuss | EMD FHL1 LMNA | XL AD AR | Contracturas articulares Niveles séricos de CK elevados Debilidad muscular en infancia / adultez temprana | Enfermedades del sistema de conducción y / o arritmias son frecuentes. |
| Hemocromatosis hereditaria | HFE | AR | Cirrosis Diabetes Pigmentación hipermelanótica Aumento de los niveles séricos de hierro y ferritina. | Cardiomiopatía no dilatada y / o infiltrativa es más frecuente que la DCM |
| Miopatía distal de Laing | MYH7 | AD | Debilidad facial Debilidad (de inicio en la infancia) de tobillos, dedos gordos, extensores de dedos y flexores de cuello. | |
| Distrofia muscular de la cintura escapular IB | LMNA | AD | Debilidad proximal de las extremidades inferiores | Enfermedades del sistema de conducción y / o arritmias son frecuentes. |
| CMD mitochondrial | ADNmt | Mat | Fenotipos complejos que incluyen: - Glomeruloesclerosis segmentaria focal - Síndrome de Kearns-Sayre |
AD: Autosómico dominante; AR: Autosómico recesiva; XL: Ligada al cromosoma X; ADNmt: ADN mitocondrial; Mat: Herencia materna
Las pruebas genéticas se deben ofrecer a todas las personas de cualquier edad con CMD no isquémica30, incluidas aquellas con cardiomiopatía periparto o asociada al embarazo31. El propósito de establecer un diagnóstico molecular de CMD es informar la evaluación de riesgos de los familiares del paciente.
Se han informado variantes patogénicas, probablemente patogénicas y de significado incierto en más de 30 genes en el 40% -50% de los individuos con CMD familiar (cuando existen más de dos familiares de primer grado afectados)32o en casos aislados (en sólo un miembro de la familia)33. La tasa de detección de variantes patogénicas y probablemente patogénicas es de aproximadamente el 27%.34
Actualmente la realización de estudios genéticos a través de paneles multigenes han permitido el análisis en simultáneo y en paralelo de todos los genes relacionados a la CMD. Dada la complejidad de interpretar los resultados de las pruebas genéticas y sus implicaciones para el seguimiento y manejo, estos casos deben ser discutidos de forma multidisciplinaria donde se incluya la asesoría genética correspondiente.35
El screening en familiares de primer grado asintomáticos de un individuo con CMD puede permitir su detección temprana, el inicio inmediato del tratamiento y la mejora de los resultados a largo plazo.36La aclaración del estado genético de los miembros de la familia de primer grado de un individuo con CMD puede informar la indicación y la frecuencia de las evaluaciones cardiovasculares correspondientes.
Cardiomiopatía arritmogénica del ventrículo derecho
La cardiomiopatía arritmogénica del ventrículo derecho (CAVD) se define por la pérdida de miocitos y la infiltración fibrograsa en el miocardio, que se asocia con una mayor susceptibilidad a las arritmias y muerte súbita, y que representa una porción significativa de muertes súbitas en atletas y adultos jóvenes.37La CAVD se hereda típicamente bajo un patrón autosómico dominante con penetrancia reducida y expresividad variable, que afecta a los hombres con mayor frecuencia que las mujeres.38
En los adultos jóvenes, el 80% de los casos se diagnostican antes de los 40 años de edad. El diagnóstico definitivo puede ser un desafío para el clínico, debido a la superposición fenotípica con otras cardiomiopatías genéticas y adquiridas.
La CAVD se describe principalmente como una enfermedad del desmosoma, un complejo multiproteico que forma uniones de célula a célula y une filamentos intermedios de células adyacentes, estableciendo así una red intercelular funcional. Los desmosomas son especialmente prevalentes en los tejidos que están sometidos a estrés mecánico, como el músculo cardíaco y la piel, lo que explica por qué el espectro fenotípico de la CAVD abarca manifestaciones cardíacas y cutáneas. Los mecanismos moleculares de CAVD incluyen la alteración de la adhesión célula-célula y la transmisión defectuosa de la fuerza contráctil.39
La mayoría de las variantes patogénicas en CAVD están presenten en los genes JUP, DSP, DSC2, DSG2 y PKP2.40Además, se han descrito variantes homocigotas o heterocigotas compuestas en los genes DSP y JUP en pacientes con CMD o CAVD.41Se han descrito además algunos genes no desmosómicos, incluido el gen TMEM43.42El análisis de la secuencia codificante de los genes desmosomales pueden identificar una variante patogénica hasta en 50% de los individuos con CAVD, siendo el 40% de casos debido a una variante en el gen PKK2.40
Cardiomiopatía por ventrículo no compactado
La cardiomiopatía por ventrículo no compactado (CVNC) aislada, se caracteriza por una apariencia muy trabeculada del miocardio del VI. Se cree que la detención en la compactación miocárdica durante el primer trimestre del desarrollo embrionario es sería la causa, sin embargo diversos autores han propuesto que puede ser un proceso adquirido basado en observaciones de casos de pacientes que desarrollan CVNC luego de haber tenido hallazgos ecocardiográficos normales previos.43
La prevalencia real de la CVNC se desconoce, sin embargo existen diversos reportes que indican que varía de 0.014% a 1.3%.43Los pacientes con LVNC tienden a tener una enfermedad de inicio temprano, con una expresión clínica que varía de asintomática hasta una función cardíaca progresivamente deficiente, hipertrofia ventricular, aumento de eventos tromboembólicos y muerte súbita.44El ventrículo izquierdo generalmente se ve afectado, pero el 50% de los pacientes tienen además la afectación del ventrículo derecho.45
La Organización Mundial de la Salud lista a la CVNC como una cardiomiopatía no clasificada46así como lo hace la Sociedad Europea de Cardiología47. Por otro lado, la Asociación Americana del Corazón clasificó a la CVNC como una cardiomiopatía genética primaria en el año 2006.48
Se han descrito variantes en genes conocidos asociados a la CMD y CMH que codifican componentes del sarcómero (ACTC1, MYH7, MYBPC3 y TNT2)49, del disco Z (LDB3)50, de la lámina nuclear (LMNA)51, el complejo de glucoproteína asociada a distrofina (DTNA)52, así como el gen del síndrome de Bart (TAZ).53
Cardiomiopatía restrictiva
La Cardiomiopatía restrictiva (CMR) se caracteriza por una mayor rigidez de las cámaras ventriculares, aunque el grosor de la pared ventricular y la función sistólica generalmente se encuentran dentro de los límites normales. La mayoría de los pacientes con CMR desarrollan insuficiencia cardíaca, lo que conlleva a la muerte temprana.54
Diversos reportes sugieren que existe una superposición clínica entre la CMR y CMH.55Estudios recientes han identificado que las variantes patogénicas en genes que codifican a proteínas sarcoméricas, que incluyen a TNNI3, TNNT2, MYH7 y ACTC1.56. Además, se han identificado variantes sin sentido en el gen de la desmina (DES) en varias familias con miopatía relacionada con la desmina, que pueden presentarse con CMR.57
Las Guías de la Asociación Europea del Ritmo Cardíaco recomiendan las pruebas genéticas específicas de la variante genética específica en todos los miembros de la familia en riesgo luego de la identificación de una variante patogénica en el paciente. probando.58
Síndrome de QT largo (SQTL)
El síndrome de QT largo se caracteriza por una prolongación excesiva de la repolarización ventricular asociada a un mayor riesgo de taquiarritmias ventriculares, síncope o muerte súbita en pacientes con un corazón morfológicamente intacto.2,59La prevalencia estimada oscila entre 1:2500 y 1:5000. Sin embargo, debido a la probabilidad de que 2/3 de los pacientes sean subdiagnosticados y que aproximadamente entre el 10% y 35% de ellos tengan un intervalo QT corregido (QTc) normal, es posible que la prevalencia real sea mayor.21,22,60El SQTL se puede producir tanto por factores genéticos como por factores adquiridos. El SQTL congénito es una enfermedad hereditaria causada por distintas mutaciones en los genes que codifican para las proteínas de los canales iónicos transmembrana de Na+ o de K+.22
Existen dos tipos de SQTL congénitos clásicos: el síndrome de Jervell y Lange-Nielson (síndrome de QT largo y sordera) y el síndrome de Romano Ward (prolongación del intervalo QT aislado). Ambos se relacionan a episodios de muerte súbita cardíaca debidos a fibrilación ventricular o arritmias de tipos Torsades de Pointes que conducen finalmente a la muerte.61En total se han identificado hasta 15 tipos de LQTS, que ocasionan una relentización en el proceso de inactivación de la corriente de despolarización del sodio hacia el interior de la célula y retardan la corriente de repolarización del potasio hacia el exterior de la misma. Todo lo anterior causa un incremento de la despolarización y dispersión de la repolarización.62
Se calcula que la tasa de mortalidad de los pacientes no tratados con SQTL es aproximadamente de 1% - 2% al año. La incidencia de muerte súbita cardíaca varía de una familia a otra en función del genotipo específico. El registro internacional de SQTL informa que la frecuencia de eventos cardíacos fue significativamente más alta en los individuos con SQTL1 (63%) o SQTL2 (46%) comparado con pacientes con SQTL3 (18%).63Además, la media del intervalo QTc fue significativamente más prolongada en el grupo SQTL3 (510 ± 48 ms) comparado con los grupos SQTL1 (490 ± 43 ms) o SQTL2 (495 ± 43).64Además, se determinó que a pesar de que la mortalidad acumulada hasta los 40 años fue similar en los grupos de los tres genotipos, la probabilidad de morir durante un evento cardíaco fue superior en los individuos SQTL3 (20%) que la de los individuos SQTL1 y SQTL2 (4% para cada grupo).64Desde el punto de vista genético, SQTL es una afección heterogénea, que posee una penetrancia incompleta (40%).65
A pesar de que las mutaciones que afectan a los genes KCNQ1 (SQTL1), KCNH2 (SQTL2) y SCN5A (SQTL3) son responsables de más del 90% de los casos genéticamente definidos de SQTL66(Tabla 5) se han descrito también formas menos comunes (<1% de casos) con etiologías moleculares heterogéneas (AKAP9, ANK2, CACNA1C, CALM1, CALM2, CAV3, KCNE1, KCNE2, KCNJ2, KCNJ5, SCN4B, SNTA1) que se expresan finalmente como una prolongación del intervalo QT y aumento del riesgo de arritmias ventriculares letales. Existe una variabilidad significativa en las tasas de eventos en SQTL, incluso dentro del mismo subgrupo genético, lo cual conlleva a una diversidad de variables clínicas y genéticas que confieren (de acuerdo a su combinación de presentación en cada paciente) distintos tipos de riesgo para cada individuo.67,68 También, se determinó que la aparición de eventos arrítmicos durante la infancia es un factor importante de predicción del riesgo, ya que se ha reportado que las personas que tienen su primer evento en este período, tiene riesgo muy alto de presentar eventos futuros.69
El pronóstico de SQTL también está relacionado con la anomalía genética subyacente. Algunos datos provenientes del Registro Internacional de SQTL demuestran que la frecuencia de eventos clínicos, antes del inicio del tratamiento, desde el nacimiento hasta la edad de 40 años, fue significativamente mayor en pacientes con SQTL2 (46%) y SQTL3 (42%) en comparación con aquellos con SQTL1 (30%).68Se ha reportado también una mayor letalidad y menor respuesta al tratamiento para los eventos relacionados con SQTL3 que con otros subtipos.69Las variantes patogénicas que resultan en cambios de aminoácidos en regiones específicas del canal iónico también confieren un mayor riesgo de arritmia.2
Tabla 5. Genes asociados síndrome qt largo.
| Gen | Fenotipo de SQTL | % de SQTL atribuido a variantes patogénicas en este gen | Proporción de variantes patogénicas detectadas por determinada metodología | |
|---|---|---|---|---|
| Análisis de secuenciamiento | Análisis de deleciones/duplicaciones | |||
| KCNH2 | SQTL tipo 2 | 25%-30% | 97%-98% | 2%-3% |
| KCNQ1 | SQTL tipo 1 | 30%-35% | 97%-98% | 2%-3% |
| SCN5A | SQTL tipo 3 | 5%-10% | Todas las variantes reportadas hasta la actualidad | Ninguna reportada |
El diagnóstico confirmatorio de SQTL se realizará en pacientes que cumplan con los criterios clínicos de enfermedad y la presencia de una variante patogénica en cualquiera de los genes implicados identificados mediante pruebas genéticas; a través, del análisis de genes específicos, paneles multigenes y/o pruebas exómicas o genómicas más completas. Se realizará el análisis de secuenciación de algún gen en específico en el caso que la variante patógena familiar sea conocida.70
De las personas que mueren por complicaciones de SQTL, la muerte es el primer signo del trastorno en un estimado del 10% al 15%. Los estudios del registro del síndrome de QT largo que incluyen pacientes, individuos con una variante patógena (en su mayoría tratados) y también familiares que murieron repentinamente muestran una mortalidad acumulada antes de los 40 años de 6% -8% en el SQTL tipo 1, tipo 2 y fenotipos tipo 3.(71, 72) En individuos entre las edades de 0 y 18 años, aquellos con un fenotipo SQTL tipo 1, tipo 2 o tipo 3 tuvieron una mortalidad acumulada de 2%, 3% y 7%, respectivamente.71Desde los 19 a los 40 años, las tasas de mortalidad fueron del 5%, 7% y 5%, respectivamente.71Aunque. los eventos sincopales son más comunes en el fenotipo SQTL tipo 1 (63%), seguidos por el fenotipo SQTL tipo 2 (46%) y el fenotipo SQTL tipo 3 (18%), la incidencia de muerte es similar en los tres.71
Para el fenotipo SQTL tipo 1 (una variante patógena específica), se observó un aumento severo de la mortalidad durante la infancia (edades 1-19 años), para el fenotipo tipo 2, se observó un aumento de la mortalidad entre las edades 15 y 39 años, y en el tipo 3 fenotipo, se observó un aumento de la mortalidad entre las edades de 15 y 19 años.73
Algunos tipos de SQTL están asociados con un fenotipo que se extiende más allá de la arritmia cardíaca72:
El síndrome de Andersen-Tawil (SQTL tipo 7) se asocia con un intervalo QT prolongado, debilidad muscular y dismorfismo facial.
El síndrome de Timothy (SQTL tipo 8) se caracteriza por un intervalo QT prolongado y características de desarrollo neurológico de manos / pies, faciales.
El síndrome de Jervell y Lange-Nielson (JLNS), un trastorno SQTL asociado con las variantes patógenas bialélicas KCNQ1 o KCNE1, está asociado con una pérdida auditiva neurosensorial profunda.
Es importante señalar que el SQTL asociado a variantes patogénicas bialélicas o heterocigotas en dos genes diferentes (variantes patogénicas digénicas) generalmente se asocia con un fenotipo más grave con un intervalo QT más largo y una mayor incidencia de eventos cardíacos.74Hasta la fecha no se ha establecido correlaciones específicas de genotipo-fenotipo. Además, el SQTL exhibe una penetrancia reducida en relación con los hallazgos del ECG. Aproximadamente el 25% de las personas con una variante patogénica tienen un intervalo QT normal (<440 ms) en el ECG basal. En un estudio realizado por Priori, se determinó que el porcentaje de individuos genéticamente afectados con presencia de un QTc normal fue mayor en el grupo SQTL tipo 1 (36%) que en el grupo SQTL tipo 2 (19%) o tipo 3 (10%).75Además, la penetrancia de los síntomas también se reduce, al menos el 37% de las pacientes con fenotipo SQTL tipo 1, el 54% con el fenotipo tipo 2 y el 82% con el fenotipo tipo 3 permanecen asintomáticos.(76)
Síndrome de QT corto (SQTC)
El síndrome de QT corto (SQTC) es una canalopatía heredada de manera autosómica dominante que se caracteriza principalmente por un intervalo QT más corto de lo normal en el EKG (< 350 ms), arritmias auriculares y ventriculares y una predisposición a la muerte súbita cardíaca.77,78Debido a que es una condición rara, los datos acerca de su prevalencia y datos demográficos aún son limitados.
La mayoría de los pacientes con SQTC tienen una historia familiar de muerte súbita y/o FA. La edad a la aparición de manifestaciones clínicas puede ser la infancia, por lo que se ha catalogado como una posible causa de muerte súbita del lactante. La gravedad de las manifestaciones clínicas del síndrome de QT corto es muy variable, desde asintomático a la FA, síncope recurrente y muerte súbita.79
Los análisis genéticos revelan que SQTC es una enfermedad genéticamente heterogénea (Tabla 6) con variantes patogénicas de ganancia de función en el gen KCNH2, dando lugar al síndrome de QT corto de tipo 1 (SQTC1), en el gen KCNQ1 asociado al síndrome QT corto de tipo 2 (SQTC2), en el gen KCNJ2 asociado al síndrome de QT corto de tipo 3 (SQTC3) y variantes de pérdida de función con un fenotipo de Síndrome de Brugada en el gen CACNA1C , dando lugar al síndrome de QT corto de tipo 4 (SQTC4) y en el gen CACNB2 , dando lugar al síndrome de QT corto de tipo 5 (SQTC5).(80)Los datos de una cohorte de pacientes, sugieren que los portadores de mutaciones en KCNH2 pueden presentar un intervalo QT más corto.(81)
Tabla 6. Genes asociados síndrome qt corto.
| Subtipo | Modo de herencia | Gen | Efecto neto de la mutación |
|---|---|---|---|
| SQTC1 | AD | KCNH2 | Ganancia de función de IKr |
| SQTC2 | AD | KCNQ1 | Ganancia de función de IKs |
| SQTC3 | AD | KCNJ2 | Ganancia de función de IK1 |
| SQTC4 | AD | CACNA1C | Pérdida de función de ICa. |
| SQTC5 | AD | CACNB2 | Pérdida de función de ICa. |
Como la mayoría de las canalopatías heredadas, el SQTC es genéticamente heterogéneo con un modo de herencia autosómico dominante. Tanto las mutaciones de ganancia de función en los canales de potasio como la pérdida de función en los canales de calcio se han implicado en la base genética de la enfermedad.81Hasta la fecha se han identificado variantes patogénicas en cinco genes diferentes que son responsables los subtipos del síndrome que van desde SQTC1 hasta SQTC52.
El diagnóstico de SQTS debería ser considerado en los pacientes que cuenten principalmente con un estudio de EKG que revele un intervalo QT corto, antecedentes familiares de SQTS o de muerte súbita antes de los 40 años de edad.82
Síndrome de Brugada (SBr)
El síndrome de Brugada (SBr) también conocido como el síndrome de bloqueo de rama derecha, elevación persistente del segmento ST y muerte súbita es una canalopatía hereditaria descrita por primera vez en el año 1992, que se caracteriza principalmente por un patrón electrocardiográfico característico en las derivaciones precordiales derechas y la predisposición a presentar arritmias ventriculares y muerte súbita cardíaca.83Se considera causante del 4% - 12% de todas las muertes súbitas y hasta de un 20% de las muertes súbitas que acontecen en pacientes con corazones estructuralmente normales.(84
Se ha determinado que el SBr tiene una prevalencia de 5 en cada 10.000 habitantes, aunque posiblemente esta cifra subestime la prevalencia real dado a que los pacientes pueden presentar formas silentes de la enfermedad.(85En ciertas poblaciones del sudeste asiático como en Filipinas, Japón y Tailandia se considera un problema de salud endémico.(85
Si bien el SBr se transmite bajo un patrón de herencia autosómico dominante84, la penetrancia clínica estimada basada en el análisis de individuos portadores de variantes patogénicas del canal de sodio es del 16%.84A pesar de que hasta el momento se han identificado varios genes causantes del SBr, en la mayoría de los casos (~ 65%) no se identifica ninguna mutación genética.(2
En el año 1998, fueron identificadas las primeras variantes patogénicas relacionadas con el SBr en el gen SCN5A (locus 3p21), el cual codifica para el canal de sodio cardiaco.(86La selección de dicho gen en un pequeño número de familias e individuos con fibrilación ventricular idiopática y un EKG característico de SBr reveló variantes de sentido erróneo, cambio de marco y empalme. Hasta en un 28% de pacientes con pruebas genéticas positivas se encontraron variantes patogénicas de pérdida de función en SCN5A.86
Actualmente se han descrito en el gen SCN5A más de 100 variantes patogénicas distintas responsables de BrS cuyo efecto en todos los casos es la reducción de las corrientes transmembrana de sodio (INa), por una reducción cuantitativa o por una disfunción cualitativa de los canales.85,86En este subtipo de SBr (SBr1), los hallazgos clínicos que se correlacionan con una variante patogénica son: un intervalo PR más largo en el ECG en reposo y un aumento exagerado en la duración del QRS con agentes bloqueadores del canal de sodio.(87Los antecedentes genéticos parecen jugar un papel muy importante en la expresión fenotípica de la enfermedad en el ECG.(88
Las variantes patogénicas en otros genes también se han relacionado, con menor frecuencia, con la ocurrencia del SBr. Las variantes en el gen GPD1L (SBr2) fueron identificadas inicialmente mediante un análisis de vinculación en una numerosa familia89y posteriormente en víctimas del síndrome de muerte súbita infantil.(90Por otro lado, en un grupo de pacientes con SBr asociado con intervalos QT cortos, el cribado del gen candidato reveló mutaciones de pérdida de función en CACNA1C dando lugar al síndrome de Brugada de tipo 3 (SBr3) y en CACNB2, dando lugar al síndrome de Brugada de tipo 4 (SBr4).(90Otros genes relacionados a SBr menos frecuentes (<1% de casos) son: SCN1B, KCNE3, SCN3B, HCN4.(91
En los pacientes con un patrón Brugada positivo en el ECG puede ser útil la detección genética dirigida al gen SCN5A. En el caso de hallarse alguna variante patogénica, se recomienda realizar pruebas específicas de mutación en los familiares de primer grado.(82. El hallazgo de una variante genética es un indicador de potencial desarrollo del fenotipo clínico de la enfermedad. Estos pacientes deben ser monitoreados cuidadosamente con el fin de identificar la aparición espontánea del patrón Brugada tipo 1, la aparición de síntomas clínicos, o bien una combinación de ambas.(2
Taquicardia ventricular polimórfica catecolaminérgica (TVPC)
La taquicardia ventricular polimórfica catecolaminérgica (TVPC) es una forma altamente letal de la enfermedad arritmogénica hereditaria caracterizada principalmente por la ocurrencia de arritmias ventriculares polimórficas en presencia de producción de catecolaminas (a través del ejercicio físico y/o estrés) y en ausencia de cardiopatía estructural.92
La TVPC es una entidad infrecuente (1: 10.000 habitantes), pero muy importante, debido al alto riesgo de muerte súbita que supone.(92Esta cifra podría estar subregistrada debido a que el hecho de no identificar ninguna mutación genética podría llevar a diagnósticos erróneos tales como fibrilación ventricular idiopática.(93En ausencia de tratamiento adecuado se calcula que fallecerán súbitamente hasta el 50% de los pacientes afectados antes de los 20 años de edad. La edad promedio del debut de la enfermedad oscila entre los 6 y 10 años, aunque se han reportado también casos esporádicos alrededor de los 30 años.(93
La TVPC debe sospecharse en todo paciente joven, en especial niño o adolescente, que presente síncopes relacionados con el ejercicio físico o el estrés emocional, que no tenga cardiopatía estructural y que su electrocardiograma muestre un intervalo QT normal.(93En el año 2001 Priori y Lahat descubrieron la causa genética de la enfermedad identificando dos variantes de la misma95, una heredada bajo un patrón autosómico dominante asociada a variantes en el gen que codifica para el receptor cardíaco de rianodina (RyR2) y otra heredada bajo un patrón autosómico recesivo asociada con mutaciones homocigotas en el gen que codifica para la isoforma cardíaca de calsequestrina (CASQ2). Hoy en día es posible confirmar el diagnóstico a través de la realización de distintas pruebas genéticas en los genes que se saben asociados a la enfermedad.(96,97)
Posteriormente, se identificaron mutaciones en el gen de la calsequestrina 2 (CASQ2) en un paciente con un patrón de herencia autosómica recesiva de TVPC (TVPC2).95Este fue el segundo gen relacionado a la TVPC. La calsequestrina 2 es una proteína crucial en la regulación del Ca2+ intracelular. Constituye el mayor reservorio de este ion en el retículo sarcoplásmico y modula además su liberación a través del RyR2. Cuando disminuye la capacidad de la calsecuestrina 2 para almacenar Ca2+, se incrementan las concentraciones de este ion libre en el espacio intercelular, hecho que determinará la prematura activación del RyR2 que dejará escapar más iones Ca2+ durante la sístole.(96
Aproximadamente un 30% - 40% de los pacientes con TVPC cuenta con algún antecedente familiar de muerte súbita; hecho que respalda el origen genético de esta enfermedad.(93Hasta la fecha, se han caracterizado 130 mutaciones en el RyR2, que representan entre el 50-60 % del total de las identificadas en la TVPC.(96Estas exhiben un patrón de herencia autosómica dominante, una penetrancia del 83 % y condicionan la TVPC de tipo 1.(93Los casos de pacientes con TVPC por variantes patogénicas en el gen CASQ2 que condicionan al TVPC de tipo 2, representan solo el 5% de todos los casos. Se heredan bajo un patrón autosómico recesivo98, debido a variantes patogénicas homocigotas u heterocigotas compuestas en el gen CASQ2 y posee una penetrancia del 78%.99
Se han identificado también alteraciones en otros genes a través del análisis de ligamiento del genoma completo que incluye a los genes de la calmodulina 1 (CALM1) en el origen de la TVPC de tipo 4 y al gen de la triadina (TRDN) como una causa de CPVT de tipo 5.(100) Finalmente, SCN5A se ha implicado en las arritmias ventriculares polimórficas inducidas por el ejercicio, con la reciente identificación de una nueva mutación sin sentido en una región altamente conservada del gen en una familia numerosa con un fenotipo CPVT.
Debe sospecharse de TVPC en todo paciente joven (principalmente niño/adolescente) que presente síncopes relacionados con el ejercicio físico, estrés emocional, en ausencia de cardiopatía estructural y con un EKG sin alteraciones (intervalo QT normal).93
El diagnóstico diferencial debe realizarse con otras enfermedades cardíacas hereditarias susceptibles de producir arritmias ventriculares malignas por el ejercicio y/o estrés emocional como el síndrome de QT largo (SQTL1, SQTL 2 y SQTL 3), ya que ambas condiciones comparten características clínicas tales como edad de debut, ausencia de cardiopatía estructural, antecedentes familiares de síncope o muerte súbita, aparición de síntomas relacionados al estrés físico o emocional y el carácter polimórfico de las arritmias ventriculares.(93La mayor diferencia entre ambas es el intervalo QT prolongado en SQTL, aunque un intervalo QT normal no excluye el diagnóstico de SQTL (5-10 % de portadores de variantes genéticas en el SQTL muestran un intervalo QT normal).
Las pruebas genéticas se recomiendan como parte de la evaluación de un individuo con taquicardia ventricular inducida por ejercicio o polimórfica (documentadas en el ECG), y cuando estas arritmias ocurren en el contexto de estrés emocional.100) También pueden considerarse en la evaluación de un paro cardíaco en el contexto de esfuerzo o estrés emocional. La secuenciación del gen RyR2 se recomienda en casos esporádicos o cuando la historia familiar sugiere una herencia autosómica dominante.100 En los casos esporádicos en que exista un historial familiar de consanguinidad o cuando se sospecha de un patrón de herencia autosómico recesivo se recomienda la secuenciación del gen CASQ2. Las variantes patogénicas en el gen CASQ2 se han identificado en sólo 1% - 2% de todos los pacientes con TVPC.100
CONCLUSIONES
El rol de los estudios genéticos en la estratificación de riesgo de muerte súbita, se basa en el estudio del paciente con sospecha clínica de un trastorno cardíaco de origen genético. Las pruebas genéticas son útiles para el diagnóstico y manejo clínico del paciente, además el asesoramiento oportuno permite la identificación de las familias en riesgo para establecer medidas de seguimiento y de prevención de forma personalizada, basada en las guías clínicas e información científica relevante.
Si bien los avances tecnológicos nos permiten actualmente tener a disposición estudios genéticos a través de paneles multigenes a través del secuenciamiento de próxima generación (NGS, next generation sequencing), y a su vez permitir la identificación de nuevas variantes asociadas a la enfermedad, no todos los pacientes tienen el acceso hospitalario a estos análisis. Una limitación de esta revisión fue no contar con una base de datos nacional sobre estudios moleculares de pacientes con enfermedad cardiaca de origen genético, esto podría deberse a que no se realizan en ningún laboratorio del país, el costo para su realización en el extranjero no podría ser cubierto por los pacientes o que no se han realizado publicaciones sobre este tema.

