











 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La displasia tanatofórica (DT) es una displasia esquelética severa y está causada por variantes patogénicas en el gen FGFR3 (receptor 3 del factor de crecimiento de fibroblastos). Las variantes patogénicas más comunes se dan en el dominio extracelular ya sea por una sustitución de cisteína por arginina en la posición 248 (Arg248Cys) o sustitución de cisteína por tirosina en la posición 373 (Tyr373Cys), mientras que para la displasia tanatofórica tipo 2 es (Lys650Glu). La DT tipo I presenta micromelia con arqueamiento femoral y craneosinostosis de grado variable, mientras que la DT tipo II se caracteriza por micromelia con fémures rectos, presencia de craneosinostosis de grado moderado a severo y deformidad de cráneo en “trébol”1
En relación a las displasias esqueléticas en Latinoamérica el estudio más grande muestra una prevalencia de 3.2 casos en 10.000 recién nacidos vivos (RNV)2. Mientras que la displasia tanatofórica es más rara aún con 0.37 casos en 10.000 RNV3.
Hasta la fecha en Perú no existen casos publicados de displasia tanatofórica tipo 1, con confirmación por estudio de panel de displasias esqueléticas de forma temprana y a su vez reportada a la comunidad científica.
Presentamos un caso de displasia tanatofórica tipo I que fue diagnosticada por la detección de la mutación del gen FGFR3 en la región central del Perú y se cuenta con el consentimiento informado para los fines exclusivamente médicos de publicación.
Reporte de caso:
Neonato varón de 38 semanas, nacido de cesárea electiva debido a malformaciones esqueléticas y polihidramnios diagnosticadas por ecografía a las 36 semanas (estudios ecográficos anteriores de forma particular, sin aparentes alteraciones). La edad materna fue de 40 años y la edad paterna de 46 años, G4P3003, VRDL: no reactivo, VIH: no reactivo y controles prenatales completos en centro de salud rural.
A la ampliación de anamnesis no se encontró antecedentes de endogamia, uso de drogas, alcohol o tóxicos. Asimismo, los padres no mostraban enfermedades actuales y tampoco hubo antecedentes familiares de enfermedades congénitas.
El paciente al nacer requirió estimulación táctil, aspiración de secreciones y 01 ciclo de ventilación a presión positiva con pieza en T (Neopuff®), score APGAR: 61-85, gasometría arterial de cordón umbilical: pH: 7.18, PaO2: 55.1 mmHg, PaCO2: 52 mmHg, HCO3: 15.8 mmol/L, EB: -9.8 mmol/L, lactato: 4.5 mmol/L, SatO2: 85%, escala Silverman - Anderson: 5 puntos, con ello se decide pasar a soporte CPAP (nasal) mejorando parcialmente la dificultad respiratoria y SatO2: 92%. A las 8 horas se cambia dispositivo a cánula de alto flujo con respuesta aceptable.
En la evaluación antropométrica se encontró un peso: 2710g, talla: 39 cm, perímetro cefálico: 36 cm, perímetro toráxico: 29.5 cm. Además, dismorfismo craneofacial, macrocefalia relativa, cuello corto, tórax estrecho, talla baja, piel en acordeón, acortamiento rizomélico de las cuatro extremidades, braquidactilia e hipotonía generalizada (Fig. 1).
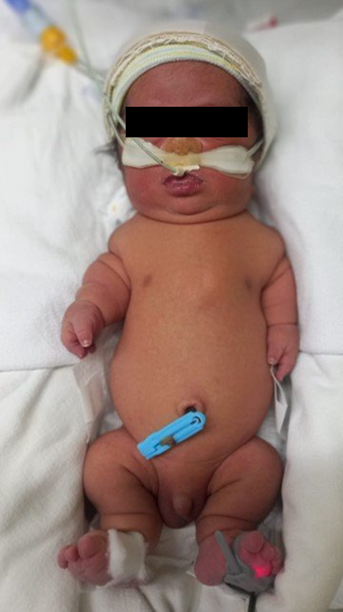
Fig.1.- Fotografía de cuerpo completo a las 24 horas de vida: Dismorfismo craneofacial, macrocefalia relativa, cuello corto, tórax estrecho, acortamiento rizomélico, braquidactilia.
El estudio radiográfico mostró una base craneal pequeña, platispondilia, arcos costales en disposición de “campanario” en relación a un tórax estrecho, ilión hipoplásico y arqueamiento femoral, con el clásico hallazgo de “receptor de teléfono” (Fig. 2).

Fig. 2 “Babygrama”, muestra hallazgos compatibles con displasia tanatofórica tipo 1 (DT 1), como: tórax en “campana”, platispondilia, hipoplasia de huesos pélvicos y fémures curvos.
Por otro lado, se realizó un panel genético para displasias esqueléticas que incluía 358 genes, identificándose 01 variante patogénica: en el gen FGFR3, variante c.1108G>T (p.Gly370Cys), (NM_000142.4).Incidentalmente se encontró en el gen KIAA0753 (NM_014804.2), una variante c.2656C>T (p.Arg886*) que está asociada a ciliopatías esqueléticas autosómicas recesivas. Esta variante no ha sido reportada en la literatura en individuos afectados con condiciones relacionadas con KIAA0753 (Fig. 3)
La evolución clínica mostró deterioro progresivo en el tiempo y pese a los diferentes dispositivos se soporte oxigenatorio y ventilatorio, falleció a los 16 días debido a un paro respiratorio secundario a la hipoplasia pulmonar y estrechez considerable de la caja toráxica.
DISCUSIÓN
La displasia tanatofórica (DT) es un desorden autosómico dominante, típicamente causada por una variante patogénica de novo, en ella tenemos un subgrupo relacionado a las condrodisplasias esqueléticas las cuales se dividen en 02 tipos (aunque no siempre guardan una correlación con el gen hallado); éstas son la DT tipo I con fémur curvo en “receptor de teléfono”, platispondilia, por lo general cráneo normal y DT tipo II con fémur recto y cráneo en “trébol”; fenotípicamente muestran un acortamiento de todas las extremidades, pliegues cutáneos redundantes, puente nasal deprimido, abdomen grande y tórax estrecho que va asociado a un pobre desarrollo pulmonar. En relación a la DT1 ésta al igual que la DT2, es causada por una mutación heterocigota en el gen que codifica el receptor 3 del factor de crecimiento de fibroblastos (FGFR3; 134934) en el cromosoma 4p16, teniendo por código OMIM #1876001,4
En cuanto al diagnóstico, se recomienda que sea precoz y de forma antenatal dado su pronóstico y desenlace; el método usado es el ultrasonido obstétrico y los criterios a considerar son: micromelia rizomélica severa con arqueamiento longitud de extremidades inferiores al percentil 3 (p3) respecto a la edad gestacional; tórax hipoplásico determinado por una circunferencia cardiaca superior al 60% de la circunferencia toráxica.6
Otros hallazgos referidos a las etapas de la gestación describen que en el primer trimestre entre la semana 12 y 14 se aprecia un incremento de la translucencia nucal y acortamiento de huesos largos, mientras que en el segundo trimestre es posible identificar la deficiencia de crecimiento de extremidades por debajo del percentil 5 (p5). Es posible también observar platispondilia, ventriculomegalia, cavidad torácica estrecha con costillas cortas, polihidramnios y arqueamiento femoral, sobretodo en la DT tipo I. Mientras que el cráneo en trébol y la craneosinostosis que afectan las suturas coronal, lambdoidea y sagital dando al cráneo un aspecto trilobulado o también denominado “Kleeblattschädel” se presentan más en la DT tipo II.1,5,7
Sin embargo, no siempre resulta fácil la evaluación de los hallazgos antes descritos es por ello, que ante la duda se recomienda complementar el estudio ecográfico usando índices como la relación: (diámetro biparietal / longitud de fémur), siendo ésta una herramienta útil y objetiva para detectar con mayor precisión las displasias tanatofóricas, sobre todo en etapas tempranas de la gestación.8,9
Otros hallazgos ecográficos, como la displasia del lóbulo temporal podrían fortalecer la sospecha diagnóstica de estar frente a un caso de DT; mientras que hallazgos morfométricos postnatales podrían aplicarse a la búsqueda ecográfica antenatal de características de DT y así optimizar la especificidad del ultrasonido obstétrico10
Las DT son un desorden de herencia autosómica dominante, esporádicos con penetrancia al 100% y de baja recurrencia; la mayoría de los casos sólo esta reportado en los afectados, siendo los padres sanos. Excepcionalmente, se ha descrito un caso de mosaicismo en una persona adulta con extremidades asimétricas, displasia congénita de cadera, áreas focales de arqueamiento óseo, húmero en “S”, acantosis nigricans extensa y óbito a las 30 semanas de una gestación con displasia esquelética severa e hipoplasia pulmonar.11
No debemos olvidar que el gen FGFR3, es responsable de una variedad de condrodisplasias, por lo que también se puede encontrar casos de hipocondrodisplasias y pseudocondrodisplasias desde grados leves hasta severos, como el síndrome de Crouzon o Pfeiffer.12
En nuestro caso, la gestante siguió sus controles obstétricos en un centro de salud rural sin alteraciones aparentes (donde no se tiene acceso pleno a personal entrenado, ni equipo ecográfico), los controles ecográficos según refiere se realizó de forma particular y sin hallazgos de anomalías, de lo cual no tuvimos evidencia física.
A las 36 semanas fue referida al hospital para completar monitoreo de gestación según normativa nacional, y es ahí donde se detecta mediante ecografía hallazgos sugerentes de DT; al nacer el bebé requirió asistencia médica inicial siendo dependiente de soporte oxigenatorio y cursando con mala evolución, llegando a fallecer a los 16 días de vida.
El diagnóstico certero se hizo mediante estudio genético e incidentalmente se detectó la coexistencia de la variante heterocigota del gen KIAA0753, la cual consideramos que no tuvo relevancia clínica al encontrarse una única mutación en dicho gen, y por ello sin consecuente expresión propia de las ciliopatias esqueléticas. Y que de otro modo hubiera resultado difícil de distinguir por el clínico entre ambas entidades.
CONCLUSIONES
En los últimos años el Perú ha sido una de las economías de la región con mayor crecimiento. Sin embargo, observamos una disociación entre ello y la posibilidad de realizar exámenes genéticos por parte de nuestros hospitales, tal como en el caso descrito. Constituyéndose así, en una barrera al acceso a estudios no sólo genéticos sino también de enfermedades raras por lo que recomendamos no sea tomado con ligereza.

