











 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Los tumores fibrosos solitarios (TFS) son neoplasias raras de células fusiformes derivadas de células mesenquimales dendríticas. Aunque afectan principalmente a la pleura visceral, también se han descrito en diversas localizaciones, incluyendo la cavidad intracraneal, las cuales representan alrededor de 0,4% de todos los tumores cerebrales primarios. Se caracterizan por altas tasas de metástasis locales, extracraneales y riesgo persistente de recurrencia incluso 10 años después de la resección inicial1,2.
El tumor afecta comúnmente a adultos en su cuarta a sexta década de vida. Según la edad de presentación, los TFS se dividen en tipo infantil (congénito) y tipo adulto. La aparición infantil es extremadamente rara y hasta ahora se han reportado menos de 20 casos. Los TFS muestran una leve predilección por el sexo masculino, los cuales se asocian a mayor riesgo de metástasis y una supervivencia de progresión libre de enfermedad más corta3,4.
En este reporte de caso, presentamos a un paciente varón de 59 años con tumor fibroso solitario intracraneal de difícil diagnóstico y la importancia de los estudios anatomopatológicos e inmunohistoquímicos para su precisión.
REPORTE DE CASO
Paciente masculino de 59 años con antecedentes de hipertensión arterial en tratamiento e infarto de ganglio basal en 2017 con secuelas (disartria, hemiparesia). Acude a consulta externa de neurocirugía (04/01/23) por presentar cefalea de 3 meses de evolución que se intensifica 2 semanas antes del ingreso, asociado a bradipsiquia. En la tomografía cerebral sin contraste (20/12/22) se evidenció una tumoración neoformativa que se extiende desde cavidad nasal, celdillas etmoidales y fosa craneal anterior, comprometiendo lóbulo frontal izquierdo con desplazamiento de línea media (Figura 1), por lo que se realizó una craniectomía exploratoria supra e infratentorial (23/01/23) donde se evidenció tumor de consistencia dura, tabicada, con abundante vascularización, adherido a línea media y techo de orbita con continuidad hacia fosas nasales y senos paranasales.
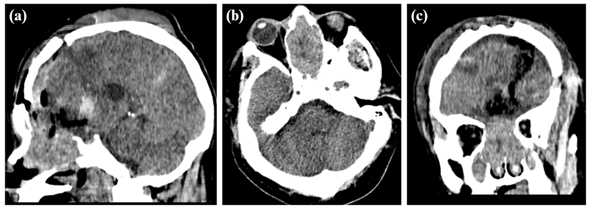
Figura 1: Tomografía cerebral sin contraste en planos sagital (a), axial (b) y coronal (c) que muestran una tumoración voluminosa expansiva en fosas nasales con mala interfase y extensión hacia nasofaringe y fosa craneal anterior, la masa condiciona remodelamiento de paredes óseas con zonas de reabsorción ósea a nivel de celdillas etmoidales y senos paranasales.
En la anatomía patológica (13/02/23) se observó tejido cerebral infiltrado de forma difusa por células tumorales en configuración sólida con núcleos ovales a alargados, pleomorfismo moderado, citoplasma de bordes no diferenciados, presencia de proliferación microvascular, índice mitótico 12/10 cap. Hallazgos compatibles con neoplasia maligna infiltrante, sugestiva de neoplasia glial de alto grado, además se sugiere estudio inmunohistoquímico para precisión diagnóstica.
Se realiza una segunda craniectomía (20/02/23) para escisión de tumor residual supratentorial donde se observa lesión tumoral en base anterior y tercio superior de cavidad nasal en relación con tumor frontonasal fibroso muy vascularizado, el cual se extrae. La anatomía patológica (Figura 2) mostró tumoración con hipercelularidad uniforme, núcleos redondos a ovalados, con pleomorfismo moderado, vasos ramificados (en asta de venado) intratumorales. Hallazgos histológicos compatibles con tumor fibroso solitario, se sugiere inmunohistoquímica.
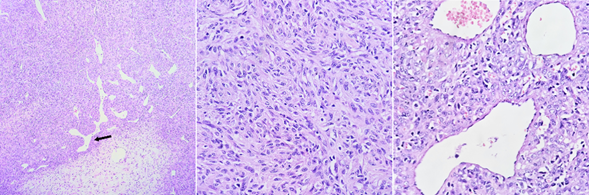
Figura 2: en las imágenes histológicas se observa a bajo aumento una proliferación neoplásica en patrón de crecimiento sólido y abundante vasculatura. Además, se observan vasos ramificados en asta de venado (flecha negra). A mayor aumento, las células son fusiformes con núcleos ovales y citoplasma eosinófilo escaso, bordes celulares indiferenciados, atipia leve, tasa mitótica variable; dispuestas alrededor de vasos sanguíneos dilatados, ramificados y congestivos, revestidos por células endoteliales planas (hematoxilina-eosina 40x).
En consulta externa por oncología (03/04/23) se observa TAC con secuela operatoria a nivel del lóbulo frontal izquierdo con masa tumoral residual de 3x3x1cm que infiltra lámina cribosa y cavidad nasal, restos hemorrágicos laminares locales, resto sin lesiones focales, no hidrocefalia, con leve desplazamiento de la línea media. Resonancia magnética (05/23) con contraste muestra tumor residual a predominio nasal/etmoidal derecho tributario a tratamiento quirúrgico. Por otro lado, la inmunohistoquímica (07/08/23) mostró resultados compatibles con tumor fibroso solitario grado 1 según la clasificación de la Organización Mundial de la Salud (OMS), donde se encontró un Ki67 con índice proliferativo <5%, CD34 y STAT-6 positivos, EMA y GFAP negativos. Además, en la histoquímica se encontró un realce de depósitos pericelulares de fibras de reticulina. Se envía caso a instituto especializado para manejo del tumor residual con radioterapia.
DISCUSIÓN
Los tumores fibrosos solitarios (TFS) son neoplasias mesenquimales poco comunes con diferenciación fibroblástica, que representan un desafío clínico debido a su comportamiento localmente agresivo, infiltración en tejidos, su tendencia a la recurrencia y su potencial para generar metástasis de manera indolente y tardía. La identificación de nuevas ubicaciones anatómicas de estas lesiones complica la diferenciación entre enfermedad primaria y metástasis5,6.
Los TFS y hemangiopericitomas (HPC) se clasificaban previamente como tumores separados. Sin embargo, después del hallazgo del gen de fusión NAB2-STAT6 en 2013, tanto el TFS como el HPC fueron tratados como una entidad única según la clasificación de la OMS para tumores de tejidos blandos. Se pueden encontrar diversas variantes asociadas a diferentes grados de agresividad, órgano afectado o metástasis7-9.
Existen 3 sistemas de clasificación (Tabla 1) siendo la más actualizada la de Marseille Grading System (MGS) donde el grado 1 se define por <5/10 HPF independientemente de necrosis; el grado 2 tiene actividad mitótica ≥5/10 HPF sin necrosis y el grado 3 tiene una actividad mitótica de ≥5/10 HPF con presencia de necrosis. Además, se observó que el grado de tumor, el recuento mitótico y la extensión de la resección eran marcadores pronósticos independientes de supervivencia libre de progresión10.
Estas lesiones pueden afectar tanto las meninges craneales (incluyendo ubicaciones dentro del tejido cerebral y la base del cráneo) como las meninges espinales (involucrando las raíces nerviosas). La mayoría de los tumores cursan con signos y síntomas acordes con su localización dentro del sistema nervioso central y el efecto de masa debido a su tamaño. Además, los más agresivos pueden ser propensos a la hemorragia como en otras lesiones de alto grado11,12.
Macroscópicamente, se presentan como tumores blandos y lobulados.
Tabla 1: Sistemas de clasificación de tumores fibrosos solitarios
| OMS 2016 | MGS 2012 | MGS 2019 |
|---|---|---|
| GRADO I Fenotipo TFS Alternancia de áreas hipo e hipercelulares Abundante colágeno Actividad mitótica >5/10 HPF* | MGS I Actividad mitótica ≤ 5/10 HPF* No necrosis No hipercelularidad | MGS I Actividad mitótica < 5/10 HPF* (independiente de necrosis) |
| GRADO II Fenotipo HPC Hipercelularidad Actividad mitótica <5/10 HPF* | MGS IIa Actividad mitótica ≤ 5/10 HPF* No necrosis Hipercelularidad MGS IIb Actividad mitótica > 5/10 HPF* No necrosis | MGS II Actividad mitótica ≥ 5/10 HPF* No necrosis |
| GRADO III Actividad mitótica >5/10 HPF* | MGS III Actividad mitótica > 5/10 HPF* Necrosis Hipercelularidad | MGS III Actividad mitótica ≥ 5/10 HPF* Necrosis |
*10 HPF (MGS): conteo de 10 áreas adyacentes con magnificación total de 400X (superficie total: 2.2 mm2) en áreas de mayor proliferación, según se evalúa en una lámina teñida con H-E o guiada por tinción inmunohistoquímica de Ki67 si está disponible. La clasificación de la OMS en 2026 no proporciona una definición para hipercelularidad y “10 HPF”. (Modificado de Macagno N. et al) (10)
Mientras que en la microscopía se pueden distinguir entre variantes hipocelulares, caracterizadas por disposición desordenada de células fusiformes ovoides con vasos dilatados y paredes finas en forma de “cuerno de ciervo” dentro de matriz de colágeno, y los densamente celulares, con células redondeadas u ovoides con poco colágeno y vasos menos pronunciados. La prominente lámina basal que se ilustra con tinciones de reticulina o colágeno IV, ayuda a diferenciarlos de los meningiomas. En los estudios inmunohistoquímicos a menudo son CD34 positivos y antígeno de membrana epitelial (EMA) negativos13,14.
El diagnóstico preoperatorio de TFS depende principalmente de los hallazgos de la resonancia magnética (RM), ya que estos tumores muestran alta vascularidad y posible naturaleza de fugas de los vasos, lo que los hace hiperintensos y fácilmente detectables con gadolinio en imágenes ponderadas en T1 y T2. Sin embargo, las características de imagen suelen ser similares a los meningiomas lo cual puede llevar a diagnósticos erróneos. Se ha observado que el volumen del tumor, el signo de cola dural y el análisis del histograma de mapas ADC pueden diferenciar los meningiomas de los TFS. La capacidad de la tomografía por emisión de positrones (PET) para detectar TFS es variable. y si bien destaca ciertos aspectos, no debería reemplazar la RM15-17.
Después del diagnóstico, la extirpación completa del TFS intracraneal es el tratamiento estándar de oro, seguida de radioterapia fraccionada. Se ha observado que la embolización arterial preoperatoria puede reducir significativamente el riesgo de hemorragia masiva intraoperatoria y mejorar la seguridad de las operaciones. La radioterapia adyuvante beneficia a pacientes con resección subtotal en términos de control local. En caso de recurrencia, la radioterapia estereotáctica es segura y eficaz para TFS de grado 1 o 2, más no se recomienda para el grado 3. La radioterapia de intensidad modulada se emplea con dosis según grado o presencia de tumor residual. El seguimiento requiere RM cada 3-6 meses en el primer año. Los TFS agresivos deben tener tomografías extracraneales anuales para detectar metástasis extraneurales16,18.
Con respecto a la radiocirugía gamma knife (RCGK), aún está en estudios, pero representa una herramienta razonable para tratar recurrencias focales de pequeño volumen en pacientes con TFS, debido a que reduce su tamaño. La RCGK es atractiva porque limita la administración de dosis a estructuras neurovasculares críticas adyacentes19.
Hasta ahora ningún agente quimioterapéutico ha sido aprobado por falta de efectividad. Se utilizan agentes citotóxicos tradicionales como doxorrubicina, ifosfamida y taxanos, con efectividad limitada. La asociación de temozolomida más bevacizumab es más efectiva y presenta menores efectos secundarios. Otras opciones incluyen inhibidores de tirosina quinasa como sunitinib y sorafenib20.
Las metástasis pueden ocurrir incluso después de una extirpación total, en promedio a los 7.5 años. Después de 10 años, con seguimiento adecuado, hay hasta un 70% de probabilidad de recurrencia o metástasis, comúnmente en hueso, hígado o pulmón. El pronóstico es generalmente malo, pero varios factores afectan la efectividad del tratamiento quirúrgico, especialmente el tamaño del tumor y el momento del diagnóstico20.
El pronóstico de los TFS se ha mejorado mediante un modelo de estratificación de riesgo que predice la probabilidad de metástasis. Este modelo utiliza cuatro variables: edad, tamaño del tumor, conteo de mitosis y presencia de necrosis. Con esta información, es posible diferenciar significativamente los diferentes grupos de riesgo, lo que permite una evaluación más precisa del curso de la enfermedad21.
CONCLUSIONES
La presentación de este caso de tumor fibroso solitario en cavidad intracraneal resalta la compleja identificación de esta neoplasia poco común. Su localización inusual, manifestaciones clínicas variables y su alta tendencia a la recurrencia y metástasis hacen que su manejo sea un desafío clínico.

