











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO  uBio
uBio 
 Permalink
Permalink
Introducción
A finales de 2019, se manifestó una nueva afección respiratoria de naturaleza infecciosa ubicada en la provincia de Hubei Wuhan, China (Huang et al. 2020). El brote fue denominado Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), que se transmitió rápidamente alrededor del mundo lo que provocó una emergencia sanitaria global. Por otra parte, la enzima convertidora de angiotensina 2 (ACE2), se identificó como el receptor funcional crítico del SARS-CoV-2. Además, ACE2 es bien conocida como un contrarregulador del sistema renina-angiotensina (RAS) y juega un rol importante en el sistema cardiovascular (Bian & Li, 2020).
Asimismo, la regulación positiva de ACE2 también está asociada con la mejora de la patogénesis de muchos trastornos metabólicos; esto incluye afecciones como la fibrosis hepática, enfermedad renal crónica, insuficiencia cardiaca y diabetes (Gottschalk et al., 2021). De la misma forma, mejora la tasa de filtración glomerular, reduce la hipertensión sistémica y atenúa la expresión de genes inflamatorios (Lo et al., 2015).
La enzima ACE2 es diana de la proteína Motivo de unión al receptor (RBM) en el dominio de unión al receptor (RBD) de SARS- CoV y funciona como receptor del SARS- CoV. De igual forma ocurre en el virus SARS- CoV-2 lo que permitiría su ingreso dentro del organismo hospedero. La especificidad de la proteína ACE2 como receptor es lo que determina el rango de hospederos que puedan tener los coronavirus dentro de la naturaleza. Se presume que los primeros virus de tipo SARS tienen su origen en animales mamíferos como los murciélagos, especulándose y comprobandose a nivel de ensayos de laboratorio que su rango de hospederos es amplio. Esto incluye a diferentes especies como gato, perro, pangolín, hámster chino y primates no humanoides como susceptibles debido a la presencia de homólogos de ACE2. El análisis de la proteína ACE2 permite determinar los posibles reservorios de coronavirus como SARS-CoV, SARS-CoV-2, entre otros. Puesto que se presume que esta proteína está ampliamente conservada a lo largo del proceso evolutivo y podría indicar la presencia de un amplio rango de hospederos. Además, para entender mejor la interacción entre la proteína viral y su receptor es necesario conocer sus estructuras tridimensionales, especialmente haciendo énfasis en los aminoácidos clave involucrados en la unión específica (Damas et al. 2020; Liu et al. 2020; Luan et al. 2020; Samavati & Uhal, 2020; Liu et al. 2021 y Wu et al. 2020).
El estudio enfocado a la predicción de la estructura tridimensional en proteínas tiene gran relevancia; dado que, proporciona una comprensión detallada y eficiente de sus propiedades y funciones a nivel molecular. De esta manera se puede determinar cuál es su rol biológico, al igual que las posibles interacciones que pueden darse con otras proteínas (Singh et al., 2022; Mehmood et al., 2020).
Actualmente, para poder obtener un análisis detallado se están utilizando recursos computacionales, lo que puede ser más rápido y menos costoso que llevar a cabo experimentos de laboratorio. Es por ello que, la bioinformática representa un campo avanzado en la biología molecular porque ofrece el empleo de métodos In silico para abordar cuestionamientos que resultaban desafiantes mediante enfoques moleculares convencionales. Estas metodologías han permitido desarrollar investigaciones en diversos aspectos como la predicción de epítopos para proteínas antigénicas de Mycobacterium tuberculosis, la identificación de homología con genes, el modelado de proteínas, el análisis mutacional, la evolución de genes antimicrobianos y la tasa de recombinación en genes de interés (Kumar et al., 2021).
Las metodologías de predicción estructural de proteínas son herramientas versátiles que permiten modelar estructuras inclusive en casos donde ya existe una estructura identificada mediante experimentación, los métodos in silico pueden ser utilizados para modelar los efectos de mutaciones, anticipar la localización de superficies de unión para otras macromoléculas y evaluar los impactos de moléculas pequeñas. Asimismo, estas técnicas permiten estimar las energías de enlace y prever movimientos locales y no locales en la estructura proteica (Werner et al. 2012).
Finalmente, el propósito del presente estudio fue analizar estructural y funcionalmente la proteína ACE2 en diferentes especies de vertebrados. Esto puede servir para futuras investigaciones que busquen determinar el rango de posibles hospederos del virus SARS-CoV-2 basándose en la homología de la proteína receptora ACE2.
Materiales y métodos
Recuperación de las secuencias aminoacídicas de la proteína ACE2
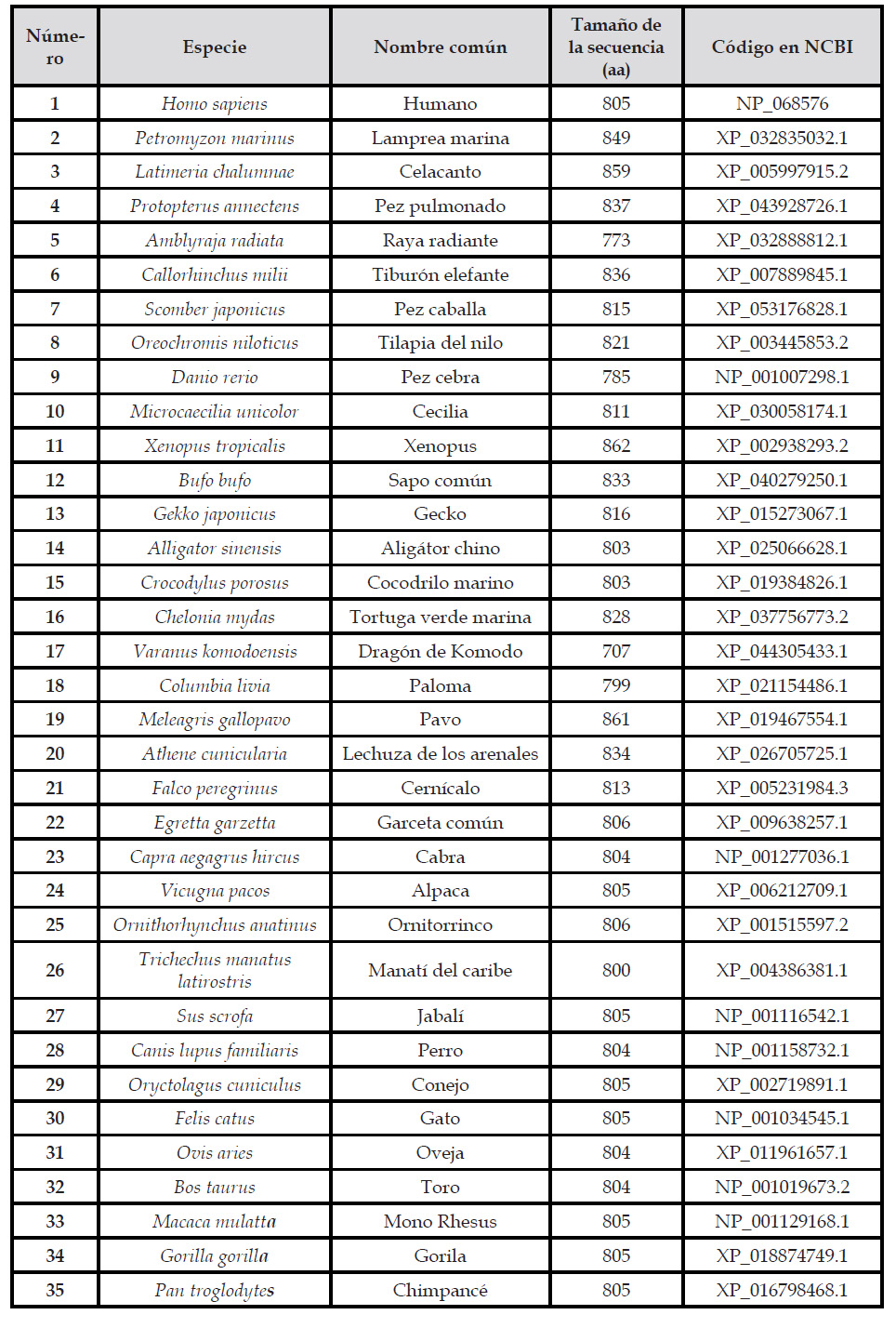
Las secuencias de la proteína ACE2 de las diferentes especies de vertebrados se recuperó de la base de datos National Center for Biotechnology Information NCBI (https://www.ncbi.nlm.nih.gov/) . Se recuperaron un total de 35 secuencias de la sección de ortólogos del gen ACE2, se tomaron como datos la especie, nombre común, tamaño de secuencia (número de aminoácidos) y los códigos de NCBI como puede evidenciarse en la Tabla Nº1. Las secuencias fueron elegidas considerando especies que pertenezcan a las diferentes clases del Subfilo Vertebrata, incluyendo a la clase Mammalia, Ave, Reptilia, Amphibia, Sarcopterygii, Actinopterygii, Chondrichthyes y Myxini. Además, se procuró que el tamaño de las proteínas no difiera mucho y que de preferencia sean determinadas con evidencia experimental. Se descargaron las secuencias en archivos de formato FASTA y fueron almacenados para posteriores análisis (Bhattacharya et al., 2018; Dutta et al., 2018; Lorrine et al., 2023; Mohanty et al., 2022; Yasin et al., 2020).
Análisis de parámetros físico- químicos de la proteína ACE2
Las propiedades físicas y químicas fueron determinadas mediante análisis computacionales utilizando el servidor ProtParam (https://web.expasy.org/ protparam/). Las propiedades evaluadas fueron Residuos, Masa molecular, Punto Isoeléctrico (pI), Índice de Inestabilidad (II), Estabilidad (tt), Índice alifático, Valor GRAVY y Coeficiente de Extinción de la proteína ACE2 de las 35 secuencias de vertebrados elegidas (Amobonye et al., 2022; Premachandran & Srinivasan, 2022; Satyanarayana et al., 2018; Singh et al., 2022 y Bhattacharya et al., 2019).
Análisis preliminar de secuencias de la proteína ACE2
El análisis preliminar de la estructura primaria busca determinar la presencia de motivos, dominios conservados, clasificación en familias de proteínas e inferir su función. En el análisis preliminar de secuencias se utilizó la herramienta bioinformática online InterPro (https:// www.ebi.ac.uk/interpro/) para determinar los dominios y clasificarlas en familias. Además, la anotación funcional y de dominios se complementa con la herramienta SMART (http://smart.embl- heidelberg.de/). Asimismo, en la búsqueda de información adicional sobre la función, clasificación estructural y de superfamilias se utilizó la herramienta CATH (https:// www.cathdb.info/) (Flores-Castañón et al., 2022; Goh et al., 2022; Lenin et al., 2023; Mukherjee et al., 2023 y Singh et al., 2021).
Predicción de estructura secundaria de la proteína ACE2
La predicción de la estructura secundaria de la proteína ACE2 de Homo sapiens y sus ortólogos se realizó con 10 secuencias seleccionadas de las 35 especies de vertebrados previamente elegidas. De las distintas clases del Subfilo Vertebrata, se consideró Petromyzon marinus de la clase Myxini, Amblyraja radiata de Chondrichthyes, Scomber japonicus de Actinopterygii, Bufo bufo de Amphibia, Chelonia mydas de Reptilia, Columbia livia de Ave y de la clase Mammalia se incluyó Canis lupus familiaris, Felis catus, Pan troglodytes y Homo sapiens.
Para su realización se utilizó el servidor PSIPRED 4.0 (http://bioinf.cs.ucl.ac.uk/ psipred/) con las opciones por defecto obteniéndose la visualización esquemática de la estructura secundaria considerando la alfa hélice, beta hoja plegada y coils. Dicho servidor online es simplificado, confiable y permite llevar a cabo la predicción de estructuras secundarias empleando redes neuronales para analizar las secuencias mediante PSI-BLAST(Kumar et al., 2021; Shrivastava et al., 2020 y Xu et al., 2021).
Predicción de estructura terciaria de la proteína ACE2
La estructura terciaria de las proteínas fue predicha utilizando el enfoque por homología mediante el uso del software MODELLER 10.4. La búsqueda de los templados se llevó a cabo usando BLAST contra la base de datos Protein Data Bank PDB (https://www.rcsb.org/) utilizando como query a las secuencias FASTA de las diferentes especies, considerando el porcentaje de identidad, query covery y eligiendo solo modelos obtenidos de análisis experimentales. Los modelos estructurales de los templados en formato PDB fueron editados utilizando PyMOL 2.5.5 para remover las cadenas adicionales como la de la proteína Spike de SARS- CoV-2 y átomos adicionales antes de su utilización. Los archivos fueron preparados adecuadamente y se generaron 5 modelos aleatorizados predichos utilizando MODELLER, almacenados en formato PDB y seleccionando el que tiene mejor valor DOPE como nuestro top-model para los siguientes pasos (Kumar et al., 2020 y Rout & Mahapatra, 2019).
Los modelos predichos deben ser evaluados y verificados para conocer su calidad y mejorarlos, para dicha labor se utilizaron las herramientas bioinformáticas disponibles en la web de UCLA-DOE LAB - SAVES v6.0 (https://saves.mbi.ucla. edu/), donde se evaluaron los parámetros que se contemplan en la herramienta PROCHECK, enfocándose principalmente en el Ramachandran Plot para el control de calidad de la estabilidad estructural (Lorrine et al., 2023; Mohanty et al., 2022 y Premachandran & Srinivasan, 2022). Aquellos modelos que no cumplieron con los parámetros evaluados, procedieron a ser mejorados mediante el refinamiento con los servidores bioinformáticos online ModRefiner (https ://zhanggroup.org/ ModRefiner/) y y GalaxyRefine (https:// galaxy.seoklab.org/cgi-bin/submit. cgi?type=REFINE). Posteriormente, se volvió a evaluar la calidad de los modelos resultantes en PROCHECK. Este proceso se realizó iterativamente hasta lograr un modelo adecuado (Amobonye et al., 2022; Desai & Chauhan, 2018). Finalmente, los modelos fueron visualizados utilizando Rasmol y PyMOL para obtener la ilustración de las estructuras más óptimas correspondientes a la estructura terciaria de cada especie.
Comparación estructural de la proteína ACE2 de de diferentes especies de vertebrados
Con el objetivo de poder comparar adecuadamente los modelos estructurales, se realizó superposición de los mismos mediante un alineamiento estructural átomo a átomo empleando el servidor web CLICK (http://cospi.iiserpune.ac.in/ click/) el cual se encarga de establecer una comparación independiente de topología de la estructura tridimensional de las proteínas, llevándose a cabo con los ajustes por defecto y mejorando la visualización de las mismas para determinar la similitud estructural (Nguyen et al. 2011).
Resultados
Parámetros físico-químicos de la proteína ACE2
Se calcularon los parámetros físico- químicos como Residuos, Masa molecular, pI, Índice de Inestabilidad, Estabilidad, Índice alifático, Valor GRAVY y Coeficiente de Extinción como se puede apreciar en la Tabla Nº2. El valor de pI para Homo sapiens es de 5.36 lo que es considerado una carga positiva en soluciones ácidas. Las demás especies analizadas presentan valores entre 5 y 6 aproximadamente. La especie con valor más bajo de pI es Sus scrofa con 4.95 mientras que el valor más alto fue de 6.03 con una especie de Meleagris gallopavo, por lo que este parámetro está altamente conservado a lo largo del proceso evolutivo. Los valores de pI ilustran cómo la composición de aminoácidos de las proteínas influye en su comportamiento en diferentes entornos de pH.
Tabla Nº2 Parámetros físico-químicos de ACE2 en especies diferentes de Vertebrados
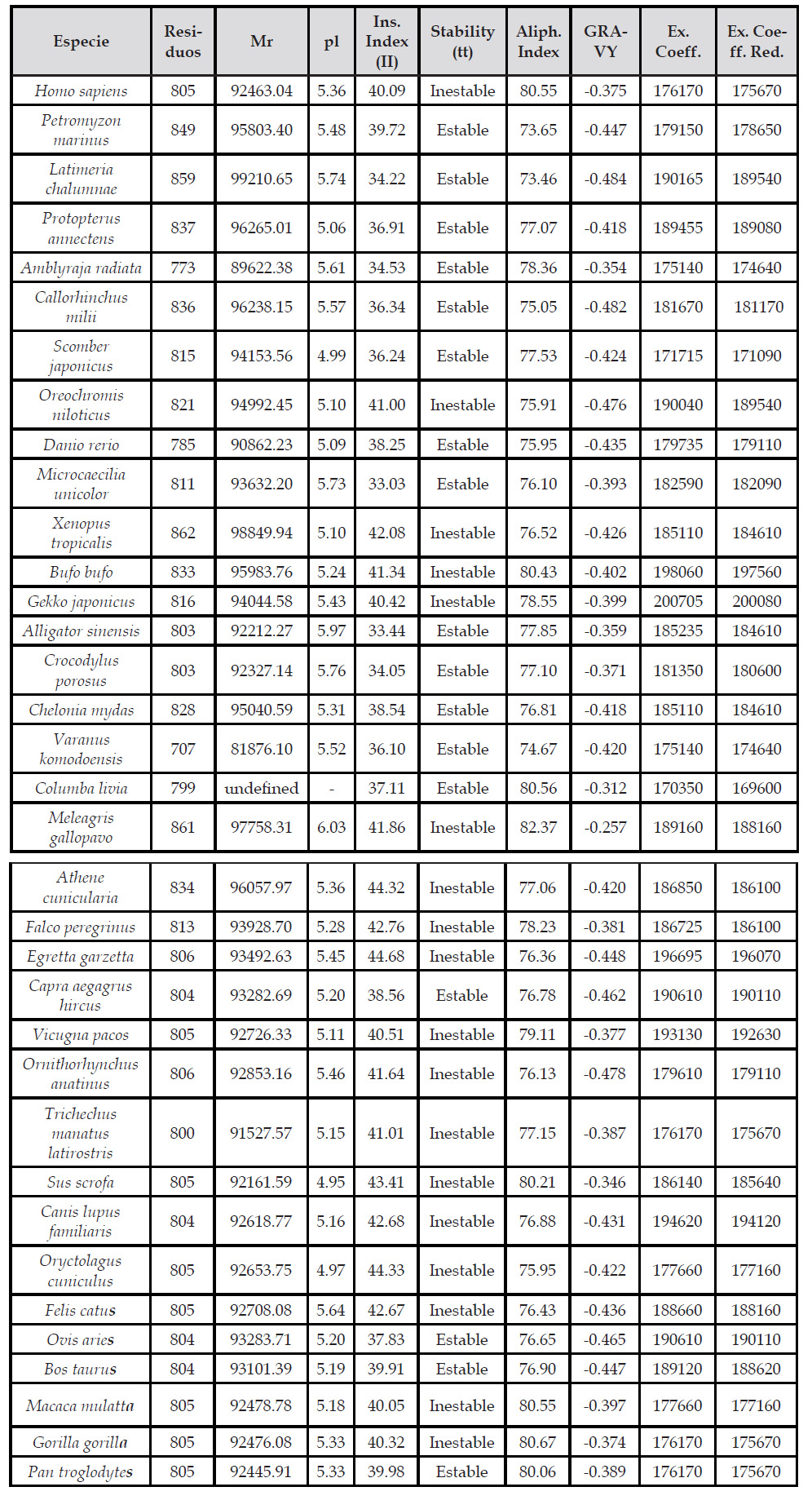
Nota: Mr: Masa molecular, pl: Punto isoeléctrico, Ins. Index (II): Índice de Inestabilidad, Stability (tt): Estabilidad, Aliph. Index: Índice alifático, GRAVY: Gran promedio de Hidrofobicidad, Ex. Coeff.: Coeficiente de extinción y Ex. Coeff. Red.: Coeficiente de extinción reducido.
Uno de los parámetros físico-químicos que tiene gran relevancia es el GRAVY (Gran Promedio de Hidrofobicidad) el cual proporciona datos sobre la capacidad de solubilidad de una proteína. La especie que obtuvo el valor más alto fue Meleagris gallopavo (-0.257) mientras que el valor más bajo perteneció a la especie Latimeria chalumnae ( -0.484), una especie bastante antigua y poco evolucionada de celacanto.
La presente investigación muestra valores de Índice de Inestabilidad en un rango de 33.03 a 44.68. Para Homo sapiens, fue de 40.09 lo que sugiere que es una proteína levemente inestable en solución y se tienen que tomar consideraciones para su almacenamiento. De la totalidad de organismos, 18 serían inestables como por ejemplo Egretta garzetta y Athene cunicularia, mientras que 17 serían estables como Microcaecilia unicolor, Alligator sinensis entre otras.
Finalmente, el rango de valores para el Índice Alifático fue de 73.46 a 82.37, siendo 80.55 para Homo sapiens lo que indica una alta termoestabilidad. La más alta termoestabilidad corresponde a Meleagris gallopavo mientras que la menos termoestable sería Latimeria chalumnae.
Análisis preliminar de la proteína ACE2 de Homo sapiens y sus ortólogos
Se ha realizado un análisis detallado utilizando herramientas bioinformáticas como INTERPRO, SMART y CATH. Donde se evidencia que pertenecen a la familia Peptidase M2 del dominio Collectrin (617- 770, 590-742, 620-773, 630-782, 623-776, 637-790, 607-760, 617-770, 616-769, 617- 770) correspondientes a las especies Homo sapiens, Petromyzon marinus, Amblyraja radiata, Scomber japonicus, Bufo bufo, Chelonia mydas, Columba livia, Canis lupus familiaris, Felis catus y Pan troglodytes respectivamente.
A su vez, también destaca por presentar como función molecular actividad metalopeptidasa y actividad peptidil- dipeptidasa en la membrana plasmática y el espacio extracelular. Sin embargo, al analizar las secuencias con la herramienta CATH, no se lograron encontrar coincidencias.
Predicción de estructura secundaria de la proteína ACE2 de Homo sapiens y sus ortólogos
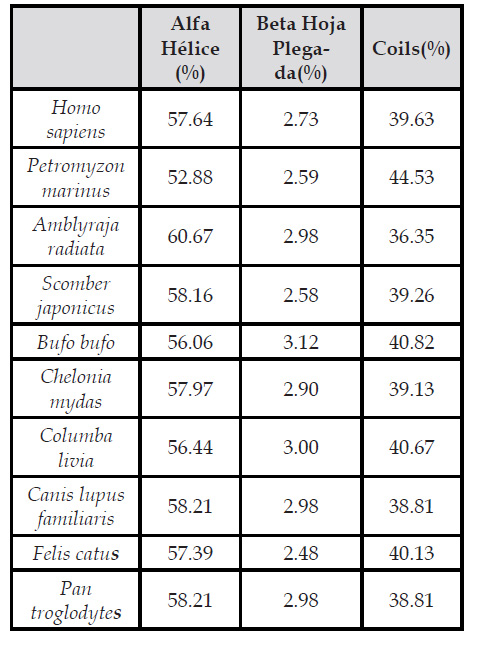
La predicción de estructuras secundarias se puede observar en la Tabla Nº3. En dicha tabla se puede apreciar el porcentaje de Alfa Hélice, Beta Hoja Plegada y Coils de cada una de las secuencias. Para Homo sapiens, la alfa hélice es la más frecuente en la conformación estructural con 57.64% mientras que la estructura menos frecuente para dicha proteína fue la beta hoja plegada con 2.73. El rango de alfa hélice varía entre 52.88 para Petromyzon marinus y 60.67 para Amblyraja radiata, lo que nos indica que es independiente del nivel evolutivo. Para la estructura beta hoja plegada los valores varían entre 2.48 para Felis catus y 3.12 para Bufo bufo, nuevamente no se puede inferir de manera directa una relación con el nivel evolutivo. Finalmente, el rango de valores para la estructura Coil fue de 36.35 para Amblyraja radiata y 44,53 de Petromyzon marinus, por lo cual no se puede inferir una asociación entre el grado evolutivo y la variación entre las diferentes estructuras secundarias de la proteína ACE2.
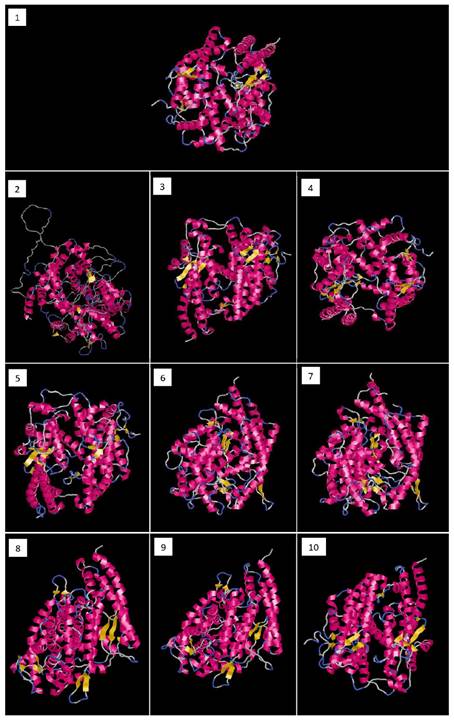
Predicción de estructura terciaria de la proteína ACE2.
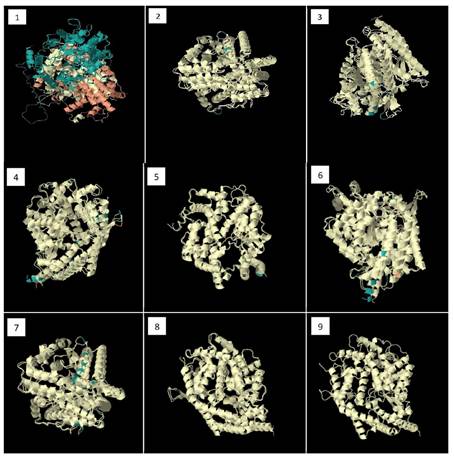
Como se puede visualizar en la figura, la estructura predominante para las 10 especies estudiadas fue la de alfa hélice; seguida de la estructura coil y la estructura beta hoja plegada que es la que se aprecia en una menor cantidad.
Un aspecto a destacar, es la divergencia observada en los modelos estructurales expuestos, teniendo una mayor pronunciación en el modelo 2 (Petromyzon marinus), con respecto a las demás especies.
Alineamiento estructural de los modelos estructurales de la proteína ACE2
Se estableció una comparación estructural entre los modelos previamente predichos para la proteína ACE2, de Homo sapiens y sus ortólogos. Esto gracias a la herramienta bioinformática CLICK, como se puede evidenciar en la figura 2. De esta manera fue posible evaluar el grado de similitud que existe entre la estructura tridimensional target con respecto a las otras especies.
Tomando en cuenta los resultados obtenidos, podemos evidenciar que existe una total correspondencia estructural entre Homo sapiens y Pan troglodytes con un porcentaje de superposición del 100%.
Por otra parte, la semejanza entre la estructura de Homo sapiens y las demás especies, también obtuvo altos porcentajes. Sin embargo, se evidenció que la alineación entre Homo sapiens y Petromyzon marinus dio como resultado un 27.30%, siendo este el menor valor obtenido.
Discusión
La proteína ACE2 desempeña un papel fundamental en las diferentes formas de vida , sobre todo en el ser humano, puesto que participa en la regulación osmótica como parte del eje Sistema Renina- Angiotensina-Aldosterona(SRA), clivando la Angiotensina II en Angiotensina 1-7 que tiene efectos importantes sobre la regulación del Sistema Cardiovascular. Además, está involucrada en la entrada del virus SARS- Cov-2 en las células hospederas, mediante su interacción con la proteína RBD del mismo.
Este virus provoca una respuesta inflamatoria, inmunitaria e infecciosa, así como también genera alteraciones en el SRA, lo que conlleva a lesiones en el pulmón y otros órganos (Bejoy et al., 2023; de Alcantara-Santos et al., 2021 y Waryah et al., 2023).
Los parámetros físico-químicos resultan determinantes de propiedades de interés en las proteínas. El número de Residuos, Masa molecular, Punto Isoeléctrico, Índice de Inestabilidad, Estabilidad, Índice Alifático, Valor GRAVY y Coeficiente de Extinción fueron determinados para las 35 secuencias de diferentes especies de vertebrados. Donde se observó que en su mayoría presentan un número de residuos parecido y cercano a la media de 805 aminoácidos, lo que estaría asociado con el grado de conservación de la estructura de la proteína. En relación al Punto Isoeléctrico (pI), se entiende como el valor de pH en el cuál las proteínas alcanzan un equilibrio entre sus cargas (generalmente entre 5 a 6), por lo que no existen fuerzas electromagnéticas de repulsión y se precipitan de la manera más adecuada (Nolsoe & Undeland , 2009). Las proteínas de ACE2 estudiadas tienen un valor de pI que oscila entre 5 y 6 lo que indica que necesitan un pH ácido para alcanzar un equilibrio entre sus cargas.
Un valor de GRAVY negativo indica que la proteína es no polar y por naturaleza su interacción con el agua es de carácter hidrofílico (Ahmad et al. 2023). En Homo sapiens, como principal secuencia objetivo, tuvo un GRAVY de -0.375 y para las demás especies evaluadas, se encontraron valores negativos en este parámetro, sugiriendo que tienen un comportamiento hidrofílico debido a la presencia de regiones aminoacídicas de este tipo. Esto indicaría que su naturaleza hidrofílica se ha conservado desde la especie que ha experimentado una menor evolución hasta la más evolucionada.
Por su parte, los Índices de Inestabilidad brindan información sobre cuál sería el mejor solvente para el almacenaje de las proteínas. Los valores inferiores a 40 indican que la proteína es estable,si los valores son más bajos presentan una mayor estabilidad mientras que valores superiores a 40 indican una inestabilidad de la proteína en solución (Grasso et al., 2016). Con respecto a este índice, se observó un comportamiento diferenciado y presentando variaciones de la estabilidad dentro de las distintas clases de Vertebrados. Esto permite inferir si las proteínas serían fácil o difícilmente conservadas en solución y no está relacionado con un comportamiento evolutivo conservado.
El coeficiente de extinción señala la cantidad de luz que puede absorber una proteína a una longitud de onda de 280 nm, lo que alude de manera implícita la cantidad de proteína presente en una solución y permite la realización de estudios cuantitativos de proteínas (Satyanarayana et al., 2018 y Dutta et al. 2018). Para el caso de Gekko japonicus, se obtuvo el valor más alto indicando una mayor presencia de proteínas en la solución, mientras que, el valor más bajo se encontró para Columba livia por lo cual no sería posible relacionarlo con el nivel evolutivo de manera directa. A su vez, el índice alifático es un parámetro físico-químico que sugiere el volumen relativo que ocupan las cadenas alifáticas laterales de aminoácidos como la valina, alanina, leucina e isoleucina en proteínas sometidas a altas temperaturas, dejando entrever su termoestabilidad.
En lo que respecta a los dominios proteicos, son importantes puesto que permiten comprender la función molecular de las proteínas y clasificarlas en familias. Para el caso de las diferentes especies de vertebrados se observó un alto grado de conservación de estos dominios ya que todas presentan el dominio colectrina y peptidasa M2, lo que las coloca en la familia de proteínas Peptidasa M2 y su función de clivaje de proteínas (Dutta et al., 2018; Lorrine et al., 2023 y Sharma et al., 2023).
Asimismo, la estructura secundaria de las proteínas resulta ser de vital importancia en el análisis de sus propiedades teniendo como base la formación de puentes de hidrógeno entre aminoácidos no contiguos. Haciendo un enfoque en las 10 especies de vertebrados seleccionadas, se observaron diferencias porcentuales entre las estructuras predichas con PSIPRED 4. La principal especie de estudio fue Homo sapiens, la cual obtuvo el porcentaje más alto de alfa hélice. que al mismo tiempo se encuentra relacionado con su presencia como proteína integral transmembrana (Feng et al. 2020 y Sang et al. 2019). Sin embargo, analizando las diferentes especies de vertebrados, se puede inferir que la composición de alfa hélice es independiente del nivel evolutivo, puesto que Amblyraja radiata tiene el valor más alto a pesar de su bajo nivel evolutivo, mientras que Petromyzon marinus presentó el valor más bajo y el menor nivel evolutivo dentro de las especies estudiadas.
Por su parte, la estructura tipo coil o giro fue la segunda más abundante que se encontró como resultado de la predicción estructural secundaria de las secuencias de proteínas. Las estructuras tipo coils resultan importantes en la estabilidad y flexibilidad así como su interacción molecular con otras proteínas (De Brevern, 2022; Hoang et al., 2019; Xu & Kennedy, 2020; Zhong et al., 2020). Al igual que con la estructura alfa hélice, no se puede inferir una asociación entre el nivel evolutivo y la composición porcentual de coils. La estructura tipo beta hoja plegada fue la menos frecuente entre todas las especies de vertebrados estudiadas. Del mismo modo, estas estructuras también son importantes por su rol en la estabilidad de las proteínas (Dhanjal et al., 2019).
Para la predicción de modelos estructurales fueron utilizadas diferentes herramientas bioinformáticas. La bioinformática estructural trata de completar el vacío de información que existe respecto a la estructura tridimensional, puesto que técnicas como la cristalografía de Rayos X, Resonancia Nuclear y Magnética y la Microscopía electrónica no se pueden utilizar tan fácilmente, ni a gran escala como para estudiar todas las secuencias de proteínas disponibles debido a su elevado costo y condiciones técnicas necesarias. Existe una alta diversidad de métodos de modelaje computacional de proteínas, algunos requieren el uso previo de bases de datos, el reconocimiento de pliegues, determinación de moldes o templados y el alineamiento comparativo de proteínas. El modelado de proteínas se fundamenta en que existe una alta similaridad entre las secuencias durante el proceso evolutivo y la homología de las diferentes proteínas, especialmente las proteínas más cercanas resultan ser las más conservadas. Entre los diferentes modelos predichos, debemos de considerar que la estructura más predominante fue la alfa hélice, seguida de los coils, lo que coincide con lo observado a nivel de la estructura secundaria predicha y resultan bastante familiares. Para comprobar la calidad de los modelos predichos se utilizaron servidores de validación que indicaron que los modelos podrían mejorarse, se utilizaron programas online de refinamiento y se volvió a verificar la calidad hasta conseguir un modelo adecuado o al menos aceptable (Dorn et al., 2014; Huang et al., 2023; Werner et al., 2012).
Para confirmar la homología estructural entre las diferentes proteínas, se realizó un alineamiento estructural con superposición de las estructuras similares. Se comprobó que la identidad entre las estructuras predichas superó inclusive el 99% para todas las comparaciones entre las diferentes especies con Homo sapiens. Solamente se encontró una marcada diferencia con Petromyzon marinus, en cuyo caso solamente hubo una identidad de 27%, lo que demuestra que esta proteína ha diferido mucho en su proceso evolutivo con todas las demás. Sin embargo, también fue el modelo predicho que presentó el mayor margen de error en su evaluación y la posible diferencia podría deberse a dificultades no superadas durante el modelaje y refinamiento de la estructura tridimensional de la proteína. Este hallazgo resulta significativo debido a que se podría deducir que existe reconocimiento grupal de ligandos basado en receptores homólogos (Kumar et al., 2021; Pollet et al., 2022; Sanchez-Pulido & Ponting, 2021).
Conclusiones
Se encontró que los parámetros físico- químicos de la proteína ACE2 entre las distintas clases de Vertebrados son muy variables. Además, se identificó que el dominio colectrina y peptidasa M2 se encontraba en todas las especies estudiadas, lo que indica su alto grado de conservación a lo largo del proceso evolutivo. También, se estableció que la estructura más predominante en los modelos tridimensionales fue la alfa hélice relacionada con su ubicación de proteína transmembrana y dicha característica está ampliamente conservada entre las diferentes clases de vertebrados.

