










Services on Demand
Journal

Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkAnales de la Facultad de Medicina
Print version ISSN 1025-5583
An. Fac. med. vol.75 no.4 Lima Oct./Dec. 2014
http://dx.doi.org/10.15381/anales.v75i4.10851
SIMPOSIO FUNCIÓN ENDOTELIAL
The vascular endothelium
El endotelio vascular
Professor Sir Salvador Moncada1
1 Director of Cancer Sciences, University of Manchester, United Kingdom.
Resumen
La investigación sobre el endotelio vascular en los últimos 40 años ha provisto ideas para entender la enfermedad vascular. Este nuevo conocimiento ha encontrado su camino en la medicina clínica. En esta revisión nos ocupamos de ciertas áreas de la investigación en las que se ha obtenido avances significativos en la prevención y el tratamiento cardiovascular, así como algunas interrogantes que aún permanecen sin respuesta.
Palabras clave: Endotelio vascular, enfermedad vascular, investigación cardiovascular.
Abstract
Over the last 40 years, research on the vascular endothelium has provided important clues for the understanding of vascular disease. This new knowledge is finding its way into clinical medicine. In this review we deal with some areas where significant advances in the prevention and treatment of cardiovascular research has been achieved and with some of the remaining questions.
Keywords: Vascular endothelium, vascular disease, cardiovascular research.
The vascular endothelium, the innermost layer of blood and lymphatic vessels, covers the whole surface of the vascular system and provides the interface between circulating blood or lymph and the vessel wall. It is one cell thick and, following its discovery in the 19th century, it was long believed to be an inert layer that merely facilitated the circulation of fluids around the body. It was Florey who, while describing his early pioneering work on the ultrastructure of the vascular endothelial cell, predicted that important discoveries could be made when pursuing the study of these cells despite the fact that the endothelium had until then been considered to be just a kind of cellophane wrapping (1). He was right.
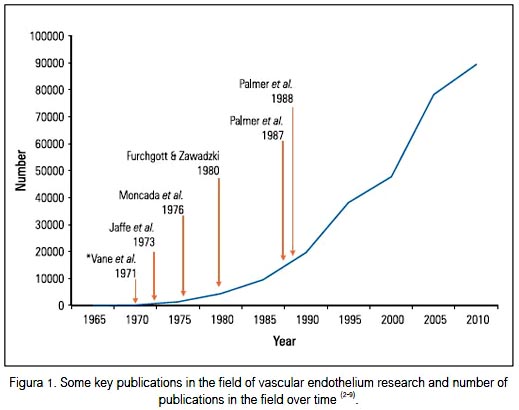
In the last 40 years, research on the vascular endothelium has been very productive and its results have greatly contributed to our understanding of the normal functioning of the vasculature as well as providing important clues for unraveling the mystery of cardiovascular disease, its origin, development, complications and its prevention or treatment. The endothelium is now considered to be an organ with significant physiological roles rather than an inert surface. Figure 1 shows the number of publications on this subject over the last four decades as well as some key discoveries that have directly contributed to the developing interest in the vascular endothelium. These include the discovery of the vasodilator prostacyclin, that of endothelium-derived relaxing factor and its identification as nitric oxide (NO). The author has described elsewhere his own contribution to this research (10).
The purpose of this brief review is to revisit some areas in which research is at present generating significant new information or where translation into clinical medicine is taking place. These include the significance of the balance between prostacyclin and thromboxane A2 for vascular homeostasis, the potential use of aspirin and related compounds in the prevention of cancer, endothelial dysfunction and the use of prostacyclin as a drug for the management of pulmonary hypertension.
The unexpected finding of the vasodilator prostacyclin while we were looking for the vasoconstrictor throm-boxane A2 in the vascular wall (10) revealed that two compounds with opposing biological functions, derived from the same precursor (arachidonic acid), are synthesized by cyclooxygenase enzymes in the platelets and the vascular wall. This led us to the hypothesis that a balance exists between the generation of these two compounds (thromboxane A2 from the platelets and prostacyclin from the vessel wall) and that this is not only important for the understanding of the homeostasis of platelet-vessel wall interactions, but also for the understanding of disease. A closelyre-lated question concerns the net effect achieved in the vasculature following treatment with aspirin and aspirin-like drugs, which have the ability to inhibit the synthesis of both prostacyclin and thromboxane A2. This has proven to be an enduring question, the answer to which is only becoming clear in the last few years.
The unique action of aspirin was unraveled in the late 1970s when it was demonstrated that the platelet cyclooxygenase, unlike that of the vessel wall, is exquisitely sensitive to aspirin and that the acetylation by aspirin of a serine residue at the active site of the enzyme is irreversible and lasts for the duration of the life of the platelets (11-13) which are unable to synthesize new proteins. This, together with the demonstration that a small dose of aspirin is more effective than a large dose in increasing cutaneous bleeding time in humans (14), led to the understanding of the now well-recognized protective effect of low doses of aspirin against vascular disease. This protective effect has been demonstrated in a large number of clinical trials in different cardiovascular conditions (15,16). The work on aspirin lent support to the hypothesis of the significance of the balance between prostacyclin and thromboxane A2, since what a low dose of aspirin achieves by selectively inhibiting generation of thromboxane A2 is to shift the balance in favor of prostacyclin.
Further support for the hypothesis came from an unexpected source. In the 1990s it was discovered that cyclooxygenase exists in two forms, one constitutive (called COX1) which generates prostaglandins for physiological functions, and a second, inducible form (called COX2) which is expressed during pathological conditions and generates prostaglandins involved in inflammation (17). Each enzyme is encoded by a different gene and their molecular structure is sufficiently different to warrant the pursuit of selective inhibitors of the COX2 enzyme. It was believed that these types of compounds would possess anti-inflammatory activity without the side effects (particularly gastric side effects) that bedevil the classical non-steroidal anti-inflammatory drugs (NSAIDS). In the event, such compounds were synthesized and the objective of achieving similar anti-inflammatory activity to the traditional aspirin-like drugs with reduced gastric side effects was achieved (18,19). However, during the development of these compounds a potential problem was identified (20-22) which was later confirmed in patients, namely that COX2 inhibitors increase the risk of cardiovascular events (18,19). Studies indicated that this serious side effect was due to inhibition of the generation of prostacyclin in the vasculature, leading to an increase in blood pressure and thus to a prothrombotic state. Over the last few years animal experiments (23,24) and clinical studies have produced overwhelming evidence in support of this suggestion, confirming that this is not a side effect related to any specific molecule but is associated with the pharmacological action of the whole class of compounds and is dependent on the strength and duration of inhibition of the synthesis of prostacyclin (19). Fittingly, a concomitant inhibition of COX1 with low-dose aspirin protects against this side effect through inhibition of the generation of thromboxane A2 (23,25).
Two problems remain to be fully clarified, the first of which is whether the generation of prostacyclin in the vasculature is due to an inducible COX2 resulting from a subliminal inflammatory condition of the vasculature, or is due to a constitutive enzyme. There is a body of evidence in favor of the latter (19). However, recent evidence indicates that it may be a mixture of the two enzymes (26,27), a fact that would be in agreement with the early observation that the concentration of 6-oxo PGF1α, the stable end product of the metabolism of prostacyclin, is elevated in patients with atherosclerosis (28).
If that is correct, then COX2 overexpression would be part of an inflammatory condition and thus a defensive mechanism. The second problem relates to the question of whether classical NSAIDs also carry the risk of cardiovascular side effects. This remains a highly controversial issue which may be resolved in further clinical trials. However, it is reasonable to assume that the cardiovascular risk of these drugs will be associated with the degree and duration of inhibition of COX2 and that the ratio between COX1 and COX2 will be determinant in their relative tendency to cause this side effect (29,30). Indeed, the use of diclofenac (which has a ratio of COX1:COX2 inhibition similar to that of the COX2 inhibitor celebrex) is associated with increased cardiovascular risk, while the use of naproxen (which is a more selective COX1 inhibitor) is not (31).The data on ibuprofen, which is also a more selective COX1 inhibitor, remains controversial (32). In summary, the concept of the balance between prostacyclin and thromboxane A2 in the homeostasis of the vascular system has been validated and its relevance in health and disease is now well understood and is guiding further development of therapies. Recently, however, a genetic variant of the gene responsible for the encoding of COX2 (PTGS2), associated with lower COX2 activity, has been identified in humans. The relationship between this condition and cardiovascular risk has so far proven to be controversial (33-36). One of the reasons for this may be that this genetic variant, although associated with a decrease in excretion of 6-oxo PGF1α, seems also to be associated with decreased concentrations of thromboxane A2; this complicates interpretation of the results using the prostacyclin/throm-boxane A2 balance hypothesis.
One of the most exciting discoveries in the use of NSAIDS has been the finding that these compounds prevent the development of different forms of cancer. This effect, which was identified some years ago in large prospective clinical trials (37,38), was later attributed to the inhibition of prostaglandin synthesis, specifically that of prostaglandin E2 (PGE2), generated by a COX2 enzyme induced by inflammation associated with pre-malignant lesions (39). This prostaglandin was believed to be responsible, at least in part, for the neo-plastic transformation through its activation of pro-survival pathways (40-43). This led to the testing of COX2 inhibitors in the chemoprevention of colorectal cancers, in which a protective action was demonstrated. These trials were, however, marred by concerns related to the potential cardiovascular side-effects of these drugs, which hampered their full evaluation (44). Studies which demonstrated that the enzyme converting PGH2 to PGE2, the socalled microsomal prostaglandin E synthase-1 (mPGE-1), is overexpressed in inflammation and couples with COX2 to enhance PGE2 generation. This has led more recently to the suggestion that selective inhibitors of this enzyme may be an important therapeutic target that will result in selective inhibition of the pathological PGE2, allowing PGH2 to be converted into the physiologically active prostaglandin, prostacyclin (45,46). Overexpression of mPGE-1 has been shown in different forms of cancers and its presence is significantly correlated with a worse prognosis, at least in colorectal cancers (47,48). Although animal studies in which deletion of this enzyme has been carried out show controversial results in relation to cancer (49,50), the development and early in vitro testing of selective inhibitors of this enzyme is proceeding (51.52), and clinical trials are likely to clarify before long the viability of this hypothesis.
The origin of the inflammatory reaction in premalignant lesions has been linked to platelet activation. Evidence for this originally came from the long- term follow-up clinical trials mentioned above in which the efficacy of aspirin as an antithrombotic agent was investigated (53-55). It was noticed that ingestion of aspirin, even at the low doses used to protect against arterial thrombosis, reduced the incidence of mortality due to cancer, particularly of those of the gastrointestinal tract. These results pointed to the platelets as a culprit (55) − a suspicion that has been strengthened by several observations including the fact that aggregating platelets can produce inflammation and induction of COX2 (56,57), and that the doses of aspirin that are protective do not reach plasma concentrations sufficiently high to inhibit COX2 and are therefore likely to be inhibiting the platelet COX1 (52,58). If these results are correct, they point towards a key where their role is now fully accepted, but also in the process of neoplastic transformation. Both effects take place via a two-step process which involves the activation of a COX1 and other pathways in the platelets, followed by the induction of COX2 in a number of cells participating in the development of the atherosclerotic plaque or the tumor.
As far as prevention or antineoplastic therapy is concerned, low-dose aspirin therefore emerges as a particularly attractive option for antithrombotic and antitumor therapy, clearly superior to the more selective COX2 inhibitors which possess cardiovascular side effects and also superior to the classical NSAIDS, none of which shares with aspirin its unique selectivity of inhibition of the platelet COX1 enzyme.
Although the idea of a dysfunctional vascular endothelium was mooted many years ago (59), only in the last 20 years has it become one of the most studied areas of vascular biology. Indeed, early detection of endothelial dysfunction is proving to be predictive of cardiovascular disease and may indicate ways of preventing its development. Endothelial dysfunction occurs in a number of conditions including hypertension, diabetes (types 1 and 2), coronary artery disease and chronic renal failure (60). It has been equated with a decrease in generation of NO by the vascular endothelium and it is likely that this may indeed be its major pathophysiological cause. However, more recently, a number of other changes have been identified which indicate that, besides a decrease in availability of NO, endothelial dysfunction also comprises an increase in vasoconstrictor, pro-inflammatory and pro-thrombotic parameters (60).
The decrease in activity of NO has been attributed largely to a decrease in its availability resulting from the interaction with oxygen-derived species, mainly superoxide anion (61,62), which may be generated by a number of enzymes including NADPH oxidase, xanthine oxidase, uncoupling of NO synthase or from the mitochondrial oxidative phosphorylation chain (63-65).
More recently, it has been suggested that increases in the concentration of asymmetric dimethylarginine (ADMA) may be involved in endothelial dysfunction. This compound was discovered some years ago to be an endogenous inhibitor of the NO synthase and shown to be increased in patients with renal insufficiency (66). Since then, evidence in favor of its role in endothelial dysfunction and in cardiovascular disease has been mounting. Indeed, an increase in plasma concentration of ADMA is associated with hypercho-lesterolemia (67), and with increased cardiovascular risk factors in patients with renal failure (68). Furthermore it is predictive of acute coronary events (69), overall mortality of patients with chronic renal failure (70), and mortality in critically ill patients (71). Two independent pieces of evidence have added support to the suggestion that ADMA plays a role in vascular disease. First, it has been shown that in some forms of vascular pathology the intracellular concentration of ADMA is elevated 3 to 9-fold over physiological concentrations; these concentrations, unlike physiological concentrations, are sufficient to inhibit NO synthase, indicating that endogenous inhibitors of NO synthesis are critical factors in vascular dysfunction following injury (72). Secondly, a genetic mutation has been identified in the enzyme dimethylarginine dimethylaminohydrolase (DDAH, the enzyme responsible for the metabolism of ADMA) in some individuals with a susceptibility to pre-eclampsia (73). In summary, although a great deal of evidence has been generated supporting the concept of endothelial dysfunction, much work is still required to clarify fully the pathophysiological mechanisms involved in this early manifestation of vascular disease. It will be important to establish whether, and to what extent, early intervention has a significant effect on the development of vascular disease.
Although the powerful vasodilator and antiplatelet effect of prostacyclin suggested early on its potential use in clinical conditions associated with thrombosis and vasoconstriction (74), its main clinical use at present is in the management of primary pulmonary hypertension (75,76), where it has been shown to improve symptoms, induce remodeling of the pulmonary vasculature and reduce mortality. The difficulties related to its intravenous usage as an unstable compound, requiring continuous administration, led to the development of different formulations of prostacyclin or its analogs for intravenous, subcutaneous and inhaled administration (75,76). In addition to the use of these compounds, two different approaches have also proven to be useful in the management of primary pulmonary hypertension. These are the use of endothelin receptor antagonists and inhibitors of the enzyme 5-phosphodiesterase to boost the effect of endogenous NO on its receptor, the soluble guanylyl cyclase. These compounds, used alone or in different combinations and schedules, have revolutionized the treatment of this complex and fatal disease to the point that the long-term management with orally-active compounds is now being investigated and prostacyclin and nitric oxide receptor agonists are at present the subject of long-term clinical trials (76-80). Furthermore, the pro-liferative nature of the disease, at least in part associated with the release of platelet-derived growth factor, has led to the development and use of different kinase inhibitors (79).
In summary, research on the vascular endothelium and in closely-related areas continues to generate a great deal of interest. As this work matures, translational developments into medicine are becoming prominent, and clear clinical benefits are being demonstrated. Almost half a century after Florey, it is still possible to predict that the endothelial cell has many secrets yet to be uncovered and that, when this occurs, further avenues for the prevention and treatment of disease will be identified.
REFERENCES
1. Florey L. The endothelial cell. Br Med J. 1966;2(5512): 487-90.
2. Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nature New Biol. 1971;231:232-5.
3. Smith JB, Willis AL. Aspirin selectively inhibits prostaglandin production in human platelets. Nature New Biol. 1971;231:235-7.
4. Ferreira SH, Moncada S, Vane JR. Indomethacin and aspirin abolish prostaglandin release from the spleen. Nature New Biol. 1971;231:237-9.
5. Jaffe EA, Nachman RL, Becker CG, Minick CR. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J Clin Invest. 1973;52:2745-56.
6. Moncada S, Gryglewski R, Bunting S, Vane JR. An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature. 1976;263:663-5.
7. Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373-6.
8. Palmer RMJ, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524-6.
9. Palmer RMJ, Ashton DS, Moncada S. Vascular endothelial cells synthesize nitric oxide from L arginine. Nature. 1988;333:664-6.
10. Moncada S. Adventures in vascular biology: a tale of two mediators. Philos Trans R Soc Lond B Biol Sci. 2006 May 29;361(1469):735-59.
11. Roth GJ, Majerus PW. The mechanism of the effect of aspirin on human platelets. I. Acetylation of a particulate fraction protein. J Clin Invest. 1975;56(3):624-32.
12. Burch JW, Baenziger NL, Stanford N, Majerus PW. Sensitivity of fatty acid cyclooxygenase from human aorta to acetylation by aspirin. Proc Natl Acad Sci U S A, 1978 Oct;75(10):5181-4.
13. Patrono C, Baigent C, Hirsh J, Roth G. Antiplatelet drugs: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest. 2008;133(6 Suppl):199S-233S. doi:10.1378/chest.08-0672.
14. O'Grady J, Moncada S. Aspirin: a paradoxical effect on bleedingtime. Lancet. 1978;2(8093):780.
15. Oates JA, FitzGerald GA, Branch RA, Jackson EK,Knapp HR,Roberts LJ 2nd. Clinical implications of prostaglandin and thromboxane A2 formation (1). N Engl J Med. 1988 Sep 15;319(11):689-98.
16. Antithrombotic Trialists' (ATT) Collaboration, Baigent C, Blackwell L, Collins R, et al. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet. 2009 MAy 30;373(9678):1849-60. doi: 10.1016/S0140-6736-(09)60503-1.
17. Masferrer JL, Zweifel BS, Seibert K, Needleman P. Selective regulation of cellular cyclooxygenase by dexamethasone and endotoxin in mice. J Clin Invest. 1990 Oct;86(4):1375-9.
18. Hinz B, Renner B, Brune K. Drug insight: cyclo-oxygenase-2 inhibitors--a critical appraisal. Nat Clin Pract Rheumatol. 2007;3(10):552-60.
19. Grosser T, Yu Y, Fitzgerald GA. Emotion recollected in tranquility: lessons learned from the COX-2 saga. Annu Rev Med. 2010;61:17-33. doi: 10.1146/annurev-med-011209-153129.
20. McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci U S A. 1999 Jan;96(1):272-7.
21. Catella-Lawson F, McAdam B, Morrison BW, Kapoor S, Kujubu D, Antes L, Lasseter KC, Quan H, Gertz BJ, FitzGerald GA. Effects of specific inhibition of cyclooxygenase-2 on sodium balance, hemodynamics, and vasoactive eicosanoids. J Pharmacol Exp Ther. 1999 May;289(2):735-41.
22. Muscará MN, Vergnolle N, Lovren F, Triggle CR, Elliott SN, Asfaha S, Wallace JL. Selective cyclo-oxygenase-2 inhibition with celecoxib elevates blood pressure and promotes leukocyte adherence. Br J Pharmacol. 2000 Apr;129(7):1423-30.
23. Cheng Y, Wang M, Yu Y, Lawson J, Funk CD, Fitzgerald GA. Cyclooxygenases, microsomal prostaglandin E synthase-1, and cardiovascular function. J Clin Invest. 2006 May;116(5):1391-9.
24. Yu Y, Ricciotti E, Grosser T, Fitzgerald GA. The translational therapeutics of prostaglandin inhibition in atherothrombosis. J Thromb Haemost. 2009 Jul;7 Suppl 1:222-6. doi: 10.1111/j.1538-7836.2009.03439.x.
25. Farkouh ME, Kirshner H, Harrington RA, Ruland S, Verheugt FW, Schnitzer TJ, Burmester GR, et al. Comparison of lumiracoxib with naproxen and ibuprofen in the Therapeutic Arthritis Research and Gastrointestinal Event Trial (TARGET), cardiovascular outcomes: randomised controlled trial. Lancet. 2004 Aug 21-27;364(9435):675-84.
26. Bishop-Bailey D, Mitchell JA, Warner TD. COX-2 in cardiovascular disease. Arterioscler Thromb Vasc Biol. 2006;26(5):956-8.
27. Caughey GE, Cleland LG, Penglis PS, Gamble JR, James MJ. Roles of cyclooxygenase (COX)-1 and COX-2 in prostanoid production by human endothelial cells: selective up-regulation of prostacyclin synthesis by COX-2. J Immunol. 2001 Sep 1;167(5):2831-8.
28. FitzGerald GA, Smith B, Pedersen AK, Brash AR. Increased prostacyclin biosynthesis in patients with severe atherosclerosis and platelet activation. N Engl J Med. 1984 Apr 26;310(17):1065-8.
29. White WB. Cardiovascular effects of the cyclooxygenase inhibitors. Hypertension. 2007;49(3):408-18.
30. Patrono C, Baigent C. Low-dose aspirin, coxibs, and other NSAIDS: a clinical mosaic emerges. Mol Interv. 2009 Feb;9(1):31-9. doi: 10.1124/mi.9.1.8.
31. FitzGerald GA, Patrono C. The coxibs, selective inhibitors of cyclooxygenase-2. N Engl J Med. 2001;345(6):433-42.
32. Coxib and traditional NSAID Trialists' (CNT) Collaboration, Bhala N, Emberson J, Merhi A, Abramson S, Arber N, Baron JA, et al., Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet. 2013 Aug 31;382(9894):769-79. doi: 10.1016/S0140-6736(13)60900-9.
33. Papafili A, Hill MR, Brull DJ, McAnulty RJ, Marshall RP, Humphries SE, Laurent GJ. Common promoter variant in cyclooxygenase-2 represses gene expression: evidence of role in acutephase inflammatory response. Arterioscler Thromb Vasc Biol. 2002 Oct 1;22(10):1631-6.
34. Cipollone F, Toniato E, Martinotti S, Fazia M, Iezzi A, Cuccurullo C, Pini B, Ursi S, Vitullo G, et al. A polymorphism in the cyclooxygenase 2 gene as an inherited protective factor against myocardial infarction and stroke. JAMA. 2004 May 12;291(18):2221-8.
35. Lee CR, North KE, Bray MS, Couper DJ, Heiss G, Zeldin DC. Cyclooxygenase polymorphisms and risk of cardiovascular events: the Atherosclerosis Risk in Communities (ARIC) study. Clin Pharmacol Ther. 2008 Jan;83(1):p. 52-60.
36. Ross S, Eikelboom J, Anand SS, Eriksson N, Gerstein HC, Mehta S, Connolly SJ, et al., Association of cyclooxygenase-2 genetic variant with cardiovascular disease. Eur Heart J. 2014 Sep 1;35(33):2242-8. doi: 10.1093/eurheartj/ehu168.
37. Thun MJ, Namboodiri MM, Heath CW Jr. Aspirin use and reduced risk of fatal colon cancer. N Engl J Med. 1991;325(23):1593-6.
38. Thun MJ, Namboodiri MM, Calle EE, et al, Aspirin use and risk of fatal cancer. Cancer Res. 1993;53(6):1322-7.
39. Sahin IH, Hassan MM, Garrett CR. Impact of non-steroidal anti-inflammatory drugs on gastrointestinal cancers: current state-of-the science. Cancer Lett. 2014 Apr 10;345(2):249-57. doi: 10.1016/j.canlet.2013.09.001
40. Plescia OJ, Smith AH, Grinwich K. Subversion of immune system by tumor cells and role of prostaglandins. Proc Natl Acad Sci U S A. 1975;72(5):1848-51.
41. Ben-Av P, Crofford LJ, Wilder RL, Hla T. Induction of vascular endothelial growth factor expression in synovial fibroblasts by prostaglandin E and interleu-kin-1: a potential mechanism for inflammatory an-giogenesis. FEBS Lett. 1995 Sep 16;372(1):83-7.
42. Sheng H, Shao J, Washington MK, DuBois RN. Prostaglandin E2 increases growth and motility of colorectal carcinoma cells. J Biol Chem. 2001 May 25;276(21):18075-81.
43. Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010 Mar;10(3):181-93. doi: 10.1038/nrc2809.
44. Garcia Rodriguez, L.A., Cea-Soriano L, Tacconelli S, Patrignani P. Coxibs: pharmacology, toxicity and efficacy in cancer clinical trials. Recent Results Cancer Res. 2013. 191:67-93. doi: 10.1007/978-3-642-30331-9_4.
45. Samuelsson B, Morgenstern R, Jakobsson PJ. Membrane prostaglandin E synthase-1: a novel therapeutic target. Pharmacol Rev. 2007;59(3):207-24.
46. Wang M, FitzGerald GA. Cardiovascular biology of microsomal prostaglandin E synthase-1. Trends Cardiovasc Med. 2010 Aug;20(6):189-95. doi: 10.1016/j.tcm.2011.04.002.
47. Nakanishi M, Gokhale V, Meuillet EJ, Rosenberg DW. mPGES-1 as a target for cancer suppression: A comprehensive invited review "Phospholipase A2 and lipid mediators". Biochimie. 2010 Jun;92(6):660-4. doi: 10.1016/j.biochi.2010.02.006.
48. Chang HH, Meuillet EJ. Identification and development of mPGES-1 inhibitors: where we are at? Future Med Chem. 2011 Nov;3(15):1909-34. doi: 10.4155/fmc.11.136.
49. Nakanishi M, Montrose DC, Clark P, Nambiar PR, Belinsky GS, Claffey KP, Xu D, Rosenberg DW. Genetic deletion of mPGES-1 suppresses intestinal tumorigenesis. Cancer Res. 2008 May 1;68(9):3251-9. doi: 10.1158/0008-5472.CAN-07-6100.
50. Elander N, Ungerbäck J, Olsson H, Uematsu S, Akira S, Söderkvist P. Genetic deletion of mPGES-1 accelerates intestinal tumorigenesis in APC(Min/+) mice. Biochem Biophys Res Commun. 2008 Jul 18;372(1):249-53. doi: 10.1016/j.bbrc.2008.05.026.
51. Leclerc P, Idborg H, Spahiu L, Larsson C, Nekhotiaeva N, Wannberg J, Stenberg P, Korotkova M, Jakobsson PJ. Characterization of a human and murine mPGES-1 inhibitor and comparison to mPGES-1 genetic deletion in mouse models of inflammation. Prostaglandins Other Lipid Mediat. 2013 Dec;107:26-34. doi: 10.1016/j.prostaglandins.2013.09.001
52. Guillem-Llobat P, Dovizio M, Alberti S, Bruno A, Patrignani P. Platelets, cyclooxygenases, and colon cancer. Semin Oncol. 2014 Jun;41(3):385-96. doi: 10.1053/j.seminoncol.2014.04.008.
53. Patrono C, Patrignani P, Garcia Rodriguez LA: Cyclooxygenase-selective inhibition of prostanoid formation: transducing biochemical selectivity into clinical read-outs. J Clin Invest. 2001;108(1):7-13.
54. Rothwell PM, Fowkes FG, Belch JF, Ogawa H, Warlow CP, Meade TW. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet. 2011 Jan 1;377(9759):31-41. doi: 10.1016/S0140-6736(10)62110-1
55. Dovizio M, Tacconelli S, Sostres C, Ricciotti E, Patrignani P. Mechanistic and pharmacological issues of aspirin as an anticancer agent. Pharmaceuticals (Basel). 2012 Dec 5;5(12):1346-71. doi: 10.3390/ph5121346.
56. Barry OP, Kazanietz MG, Praticò D, FitzGerald GA. Arachidonic acid in platelet microparticles up-regulates cyclooxygenase-2-dependent prostaglandin formation via a protein kinase C/mitogen-activated protein kinase-dependent pathway. J Biol Chem. 1999 MAr 12;274(11):7545-56.
57. Lindemann S, Tolley ND, Dixon DA, McIntyre TM, Prescott SM, Zimmerman GA, Weyrich AS. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J Cell Biol. 2001 Aug 6;154(3):485-90.
58. Dovizio M, Bruno A, Tacconelli S, Patrignani P. Mode of action of aspirin as a chemopreventive agent. Recent Results Cancer Res. 2013;191:39-65. doi: 10.1007/978-3-642-30331-9_3.
59. Stemerman MB. Vascular injury: platelets and smooth muscle cell response. Philos Trans R Soc Lond B Biol Sci. 1981;294(1072):217-24.
60. Endemann DH, Schiffrin EL, eds. Endothelial dysfunction. 2004/07/31 ed. J Am Soc Nephrol. 2004;15:1983-92.
61. Gryglewski RJ, Palmer RM, Moncada S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature. 1986;320(6061):454-6.
62. Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43(2):109-42.
63. Hamilton CA, Brosnan MJ, Al-Benna S, Berg G, Dominiczak AF. NAD(P)H oxidase inhibition improves endothelial function in rat and human blood vessels. Hypertension. 2002 Nov;40(5):755-62.
64. Landmesser U, Spiekermann S, Dikalov S, Tatge H,Wilke R,Kohler C,Harrison DG,Hornig B,Drexler H. Vascular oxidative stress and endothelial dysfunction in patients with chronic heart failure: role of xanthine-oxidase and extracellular superoxide dismutase. Circulation. 2002 Dec 10;106(24):3073-8.
65. Moncada S. Mitochondria as pharmacological targets. Br J Pharmacol. 2010 May;160(2):217-9. doi: 10.1111/j.1476-5381.2010.00706.x.
66. Vallance P, Leone A, Calver A, Collier J, Moncada S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet. 1992 Mar 7;339(8793):572-5.
67. Boger RH, Bode-Böger SM, Szuba A, Tsao PS, Chan JR, Tangphao O, Blaschke TF, Cooke JP. Asymmetric dimethylarginine (ADMA): a novel risk factor for endothelial dysfunction: its role in hypercholesterolemia. Circulation. 1998 Nov 3;98(18):1842-7.
68. Zoccali C, Benedetto FA, Maas R, Mallamaci F, Tripepi G, Malatino LS, Böger R; CREED Investigators. Asymmetric dimethylarginine, C-reactive protein, and carotid intima-media thickness in end-stage renal disease. J Am Soc Nephrol. 2002 Feb;13(2):490-6.
69. Valkonen VP, Päivä H, Salonen JT, Lakka TA, Lehtimäki T, Laakso J, Laaksonen R. Risk of acute coronary events and serum concentration of asymmetrical dimethylarginine. Lancet. 2001 Dec 22-29;358(9299):2127-8.
70. Zoccali C, Bode-Böger S, Mallamaci F, Benedetto F, Tripepi G, Malatino L, Cataliotti A, Bellanuova I,Fermo I,Frölich J,Böger R. Plasma concentration of asymmetrical dimethylarginine and mortality in patients with end-stage renal disease: a prospective study. Lancet. 2001 Dec 22-29;358(9299):2113-7.
71. Nijveldt RJ, Teerlink T, Van Der Hoven B, Siroen MP, Kuik DJ, Rauwerda JA, van Leeuwen PA. Asymmetrical dimethylarginine (ADMA) in critically ill patients: high plasma ADMA concentration is an independent risk factor of ICU mortality. Clin Nutr. 2003 Feb;22(1):23-30.
72. Cardounel AJ, Cui H, Samouilov A, Johnson W, Kearns P, Tsai AL, Berka V, Zweier JL. Evidence for the pathophysiological role of endogenous methylarginines in regulation of endothelial NO production and vascular function. J Biol Chem. 2007 Jan 12;282(2):879-87.
73. Akbar F, Heinonen S, Pirskanen M, Uimari P, Tuomainen TP, Salonen JT. Haplotypic association of DDAH1 with susceptibility to pre-eclampsia. Mol Hum Reprod. 2005 Jan;11(1):73-7.
74. Moncada S, Vane JR. Prostacyclin in PersPective, in Prostacyclin. New York: Raven Press New York. 1979:5-16.
75. Safdar Z. Treatment of pulmonary arterial hypertension: the role of prostacyclin and prostaglandin analogs. Respir Med. 2011 Jun;105(6):818-27. doi: 10.1016/j.rmed.2010.12.018.
76. Duarte JD, Hanson RL, Machado RF. Pharmacologic treatments for pulmonary hypertension: exploring pharmacogenomics. Future Cardiol. 2013 May;9(3):335-49. doi: 10.2217/fca.13.6.
77. Stamm, JA, Risbano MG, Mathier MA. Overview of current therapeutic approaches for pulmonary hypertension. Pulm Circ. 2011 Apr-Jun;1(2):138-59. doi: 10.4103/2045-8932.83444.
78. Galie N, Ghofrani AH. New horizons in pulmonary arterial hypertension therapies. Eur Respir Rev. 2013 Dec;22(130):503-14. doi: 10.1183/09059180.00006613.
79. Morrell NW, Archer SL, Defelice A, Evans S, Fiszman M, Martin T, Saulnier M, Rabinovitch M, et al., Anticipated classes of new medications and molecular targets for pulmonary arterial hypertension. Pulm Circ. 2013 Jan;3(1):226-44. doi: 10.4103/2045-8932.109940.
80. Seferian A, Simonneau G. Therapies for pulmonary arterial hypertension: where are we today, where do we go tomorrow? Eur Respir Rev. 2013 Sep 1;22(129):217-26. doi: 10.1183/09059180.00001713.
Conflicts of interest:
None declared by the author.
Correspondence:
Professor Sir Salvador Moncada
Wolfson Molecular Imaging Centre 27 Palatine Road, Manchester M20 3LJ
E-mail: s.moncada@ucl.ac.uk

